
首先QTL是数量性状位点,比如身高是一个数量性状,其对应的控制基因的位点就是一个数量性状位点,而eQTL就是控制数量性状表达位点,即能控制数量性状基因(如身高基因)表达水平高低的那些基因的位点。
数量性状基因座:控制数量性状的基因在基因组中的位置称数量性状基因座。常利用DNA分子标记技术对这些区域进行定位,与连续变化的数量性状表型有密切关系
表达数量性状基因座(expression Quantitative Trait Loci,eQTL)是对上述概念的进一步深化,它指的是染色体上一些能特定调控mRNA和蛋白质表达水平的区域,其mRNA/蛋白质的表达水平量与数量性状成比例关系。eQTL可分为顺式作用eQTL和反式作用eQTL,顺式作用eQTL就是某个基因的eQTL定位到该基因所在的基因组区域,表明可能是该基因本身的差别引起的mRNA水平变化;反式作用eQTL是指某个基因的eQTL定位到其他基因组区域,表明其他基因的差别控制该基因mRNA水平的差异。
eQTL就是把基因表达作为一种性状,研究遗传突变与基因表达的相关性: 就好像研究遗传突变与身高的相关性一样
早年可以通过同时做一个个体的SNP芯片和cDNA芯片, 在全基因组尺度研究突变与表达的相关性, 这种研究需要较多个体(例如1000个); 现在随着深度测序的出现,很多人开始用RNA-Seq在较少量个体中研究allele-specific expression,本质上就是eQTL.
简单地说, 遗传学研究经常发现一些致病或易感突变, 这些突变怎样导致表型有时候不太直观; 所以用某个基因的差异表达作为过渡: 突变A-->B基因表达变化-->表型;
GTEx的前世今生
转自https://baijiahao.baidu.com/sid=1582163111276275189&wfr=spider&for=pc
然而,事情却没有那么简单,从基因的改变到疾病等现象的出现,中间缺失了重要的一环,那就是基因的表达。也许在测序中,我们可以看到某一个基因上某一个位置的变化(比如说SNP单核苷酸变化),但是这种变化并不一定会影响mRNA的产生或者蛋白的改变。也就有可能不会影响到疾病或其他生物学过程。于是科学家想到了另一个指标——mRNA的序列数据。因为只有被表转译到mRNA上的基因,才可能进一步表达为蛋白(图1)。
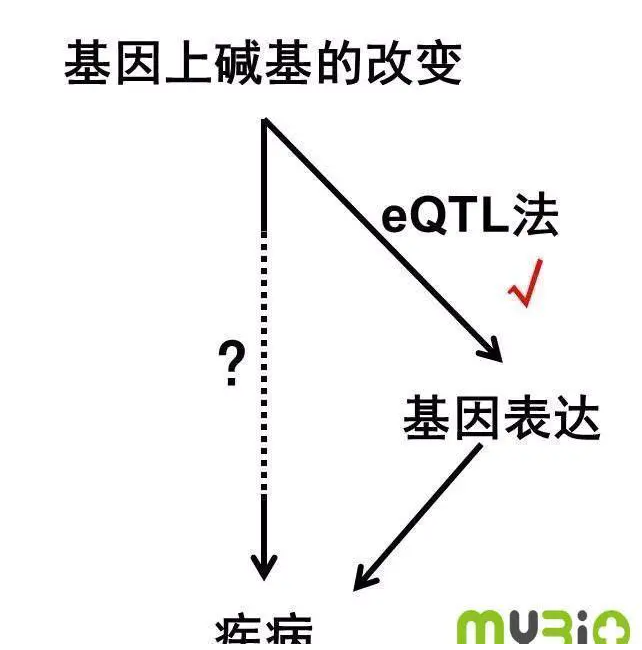
图1:eQTL是沟通基因改变与疾病的桥梁
但是要怎么搞清DNA改变是怎么影响mRNA的出现呢?这一过程被称为Expression quantitative trait loci(eQTL) 分析,目的在于得到单个DNA突变与单个基因表达量之间的相关性。与单个基因mRNA表达量相关的DNA突变,就被称为eQTL。
简单来讲,(并不简单,小编注)我们首先通过全基因组测序获得每个个体的DNA全序,然后以同种族的其他个体作为参照,标记出该个体所有的DNA变异位点, 称为SNP位点。同时,我们通过全基因组mRNA表达量测序得到该个体的特定组织样本中的基因表达量。以全部DNA变异位点为自变量,轮流以每种mRNA表达量为因变量,用大量的个体数据做样本进行线性回归,就可以得到每一个SNP位点和每一个mRNA表达量之间的关系。
GTEx是第一个收集了多个人体器官mRNA测序的数据库,并提供了跨器官的eQTL研究平台。
当前使用的GTEx v6p版本的原始数据来自于449名生前健康的遗体捐献者的44个不同的器官。图2是不同器官里面样本数的直观展示。由这个图可以看出,这一数据库中涉及的数据覆盖面非常广,数据量大,具有重要的应用潜力。
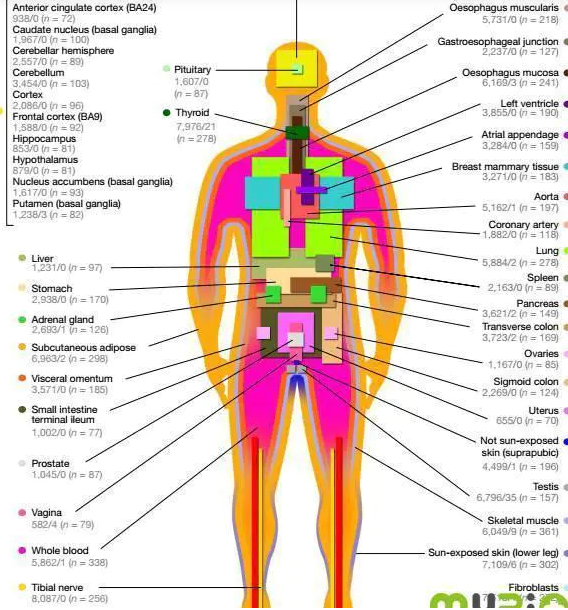
图2:GTEx 样品取材来源图示。灰色字体为 cis-eQTL 数/trans-eQTL数 (样本
3.GTEx如何一天发四篇Nature
GTEx 为挖掘器官特异的基因组数据提供了一个非常好的平台。这是目前唯一一个可以提供这些内容的数据库工具。有了这些,科学家就可以尝试回答很多问题:比如基因相互作用的网络在不同器官里会有怎么样不同的表现?不同组织中基因突变对于基因表达有哪些影响?特定染色体在不同的器官中作用有哪些?等等。也就有了其他的三篇文章。
这充分展现了该平台的强大威力!
4.核心作者专访
生息提问1:我们知道,对于大数据的分析而言,数据的质量控制与分析是非常重要的,能否详细的介绍一下你们的数据质量控制与分析过程和原理?
答:有很多文章提出,在不同的测序环境下得到的不同样本的数据存在batch effect。已知的batch effect包括测序的批次,样本的性别、年龄、祖先,以及gene的GC content等。这些因素可能同时对SNP genotype 以及gene mRNA 表达量造成影响,也就是confounder。我们不希望得到的eQTL是由于祖先的不同,或者由于性别差异,所以这些因素都被作为confounder从mRNA 表达量中通过线性回归去除。然而我们还是无法得知所有batch effect,由于我们的目的是寻找对单个基因有影响的突变,一个简单的想法是去除对mRNA 表达有广泛影响的因素, 也就是回归去除主成分。PEER就是一个可以直接用mRNA数据估计广泛因子并去除的工具。这个步骤看似简单,其实是整个分析过程中至关重要的一步,因为这个步骤直接决定了mRNA校正后的表达量。
得到校正后的数据之后,就可以对所有基因的mRNA表达量和全基因组测序得到的SNP genotype进行线性回归,即eQTL 分析。在基因组数据分析中,存在一个普遍的问题,就是基因/DNA变异的数量是样本数量的几百甚至几万倍,而同时进行的回归分析的次数就更是远超样本数量,很容易出现假阳性结果,因此我们需要对系数的p值做Bonferroni校正或者BH校正来消除多重检验的影响。
根据SNP位点(即单个DNA突变的位点)到gene的距离,eQTL 可以分为两类:cis-eQTL 和 trans-eQTL。cis-eQTL是指近距离相关的eQTL, trans-eQTL则是包括了远距离相关的eQTL。这两类eQTL对应 cis-regulation以及 trans-regulation。由于trans-eQTL的计算涉及到更大量的多重检验,因此我们采取了更严格的数据质量控制。
生息提问2:GTEx 数据是如何被用于器官特异性分析的?
答:数据收集完成后,考虑到不同器官样本里面存在重复的个体,研究人员做了meta-analysis来去除重复个体的影响,在此基础上进行了聚类分析。原文中的图2展示了用cis-eQTL 和 trans-eQTL做聚类分析的结果。可以清楚地看到,相近的器官在聚类结果中更接近。同时,cis-eQTL 比 trans-eQTL提供了更明显的分类。
一个很自然的问题就是:在不同器官里的 eQTL 有什么不同的性质?是什么因素导致一部分eQTL只存在于某些器官,而另一些则在大多数器官中存在?研究人员标记了SNP所在区域的生物学功能(promoter/enhancer/cis regulatory region),并比较了相对应器官的富集性以及不对应器官的富集性,论文中图3a,3b展示了cis-eQTL 在对应器官里更有可能在CRE中富集,图3e 则更为直接地表现了不同功能性序列的eQTL 相关性强度。
生息提问3:从找到DNA变异与mRNA表达的关联,到真正理解产生这些关联的背后机制,这中间还有多远?
答:eQTL 分析是单个SNP和单个基因表达的相关性分析,和GWAS相似,得到的结果可以为机制研究提供思路和方向。假设我们对某个基因感兴趣,想知道是什么DNA序列调控了这个基因的表达,我们可以去eQTL的list中搜索和这个基因有显著相关性的DNA序列。但是这些序列并不是直接导致基因表达变化的,主要由于两个原因:1. 染色体上靠的比较近的序列一般会同时被一代一代传下去,因此这块区域里的SNP都有固定的排列,这块区域被称为LD block。在一个LD block里面,验测出相关性的SNP可能与真正有因果性的SNP有很强的相关性,但是由于多重检验,真正有因果性的SNP可能并没有得到显著结果。2. 存在相关性并不等于直接相关,由于eQTL分析是单个基因只对单个SNP做线性回归,并没有控制其他SNP的序列,也就是说,可能这个相关性来自于另一个和基因以及这个SNP都相关的SNP。
这时候就需要其他的额外信息来帮助我们判断,哪些SNP更可能是有因果性的那个。有许多研究致力于结合其他的基因组信息,使用监督性学习或无监督学习,从存在相关性的一组SNP里面来识别真正有因果性的那个。当然,最后的因果性确认还需要严谨的生物实验来证明。
2.https://mp.weixin.qq.com/s/X6oiHtKBMBPFqncgL3Enxw
本篇给出了eQTL概况,以及三个关于eQTL的数据库,分别为Braineac,GTEx,Blood eQTL Browser

Braineac(The Brain eQTLAlmanac)由UK Brain Expression Consortium创建,他们采集134名欧洲捐献者的多个脑区的基因型与基因表达量,进行eQTL分析,建立该数据库。
http://www.braineac.org/
GTEx(The Genotype TissueExpression Project)到目前为止共收集了544名捐献者全身各个组织的基因型与基因的表达量,利用GTEx可以研究各个组织中的eQTL。
https://www.gtexportal.org/home/
Blood eQTL Browser收集了来自七个中心的共5311名被试的血液组织中的基因型及基因表达量,进行(eQTL) meta分析,并在另外的四个中心的2775被试中重复验证,他们的研究与前两个数据库相比,发现了大量的trans-eQTL。
http://genenetwork.nl/bloodeqtlbrowser/
3.又找到eQTL利用软件出结果的流程的推文,我就是资料整合专家
https://mp.weixin.qq.com/s/FaNhRYSyjLlC1hMnZJkvdA
https://mp.weixin.qq.com/s/83axhA3GgZjw4trmwhMyYw
https://www.jianshu.com/p/6e6d54d7483e
https://mp.weixin.qq.com/s/X6oiHtKBMBPFqncgL3Enxw
第三个好棒,写的超全面,也有链接到其官网数据。
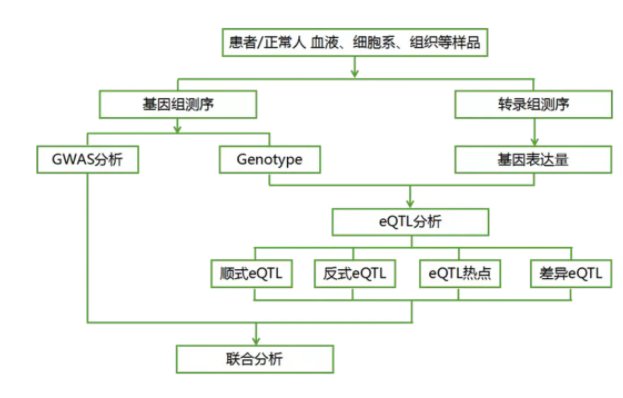
4.可做的各种结合分析:
作者:又是一只小菜鸟
链接:https://www.jianshu.com/p/2e1e9d3ccd63
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号