
转载来自https://zhuanlan.zhihu.com/p/393674599
写的非常好 怕找不到留着自己看!如果作者不同意我会删除。
前言
接下来我们要介绍的是 RNA-seq 数据的处理分析流程,根据 RNA-seq 测序技术的不同,可以分为三种:
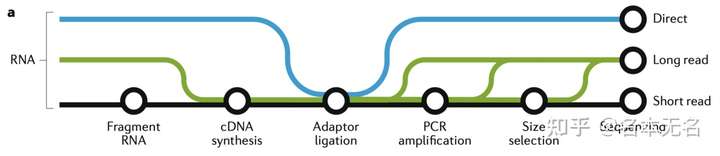 Stark et al. Nat Rev Genet(2019)
Stark et al. Nat Rev Genet(2019)
short-readlong-readdirect RNA-seq
而我们一般的 RNA-seq 测序数据分析流程算法,基本上都是基于 short-read(短读长)技术所产生的数据文件
目前,我们可以从 Short Read Archive(SRA) 数据库获取的 RNA-seq 数据中,有超过 95% 的数据是由 Illumina 公司的 short read 测序技术所产生的
其分析过程可以用下面的路线图表示
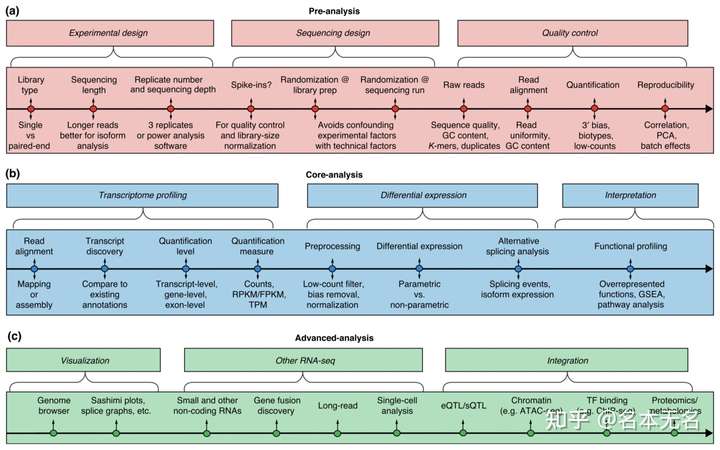 Conesa et al. Genome Biology (2016)
Conesa et al. Genome Biology (2016)
该路线图大致分为三个部分:
- 数据获取:
- 包括实验设计、测序设计以及数据下机后的
raw reads数据的质控
- 数据分析
- 在获取到干净的数据之后,可以进行
reads的比对,然后进行基因表达的量化、差异表达分析、功能富集分析等
- 高级分析
- 包括数据的可视化,其他小分子
RNA分析、融合分析以及与其他类型的数据进行整合分析等
而我们分析的起始点,是从原始数据开始的,也就是获取 raw reads 数据。通常这种高通量测序数据会保存为 FASTQ 格式的文件。
FASTQ 格式是一种以 ASCII 码字符的形式保存生物序列及其对应的每个碱基的质量的文本文件。
FASTQ 文件中每条序列(通常是一条 read)是由 4 行组成,其中:
- 第一行以
@字符开头,之后的字符为序列的标识符和描述信息 - 第二行为具体的序列
- 第三行以
+符号开头,之后可以可选地加上与第一行一样的序列标识或描述信息 - 第四行为碱基质量分数(
Phred),其字符数量与第二行相等,每个字符表示对应碱基的质量得分,例如
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65其中,碱基质量值的编码方式为
- 先将碱基错误率
P进行负对数转换,得到Q值 - 然后将
Q值加上33或64得到的值所对应的ASCII码即为碱基质量分数
例如,错误率 P = 0.01,则 Q = 20,如果是 Phred33 则对应的质量为字符 5(53),如果是 Phred64 则对应的字符为 T(84)
分析流程
1. 数据获取
一般情况下,如果自己有送样检测数据的话,测序公司会提供原始的 FASTQ 格式的数据。如果我们要使用别人文章中发表的公开数据,还需要从数据库中下载对应的数据
例如,我们从 SRA 数据中下载的原始测序文件是 sra 格式,我们需要先使用工具将其转换为 FASTQ 格式
2. 质量控制
主要在三个地方需要对数据的质量进行监控
- 获取原始数据之后
- 比对完之后
- 表达定量之后
2.1 raw read
对 raw reads 数据进行质量控制,需要分析序列的质量、GC 含量、是否存在接头、短重复序列的分布、测序错误以及 PCR 重复和污染
质控软件:
FastQC:用于分析Illumina测序平台的数据NGSQC:可应用于所有平台
一般来说,reads 的质量会朝着 3' 端递减,如果碱基的质量太低,我们需要删除它以提高比对率
FASTX-Toolkit 和 Trimmomatic 两个软件可以用于切除低质量的碱基和接头序列
2.2 比对后
reads 通常需要比对到一个参考基因组或转录组,而比对的质量是评估测序准确率和是否存在 DNA 污染的一个重要指标
比对质量通常为比对到的 reads 数占总 reads 数的比例。
例如,比对到人类参考基因组的比对质量通常需要在 70-90%,且有大量的 reads 映射到一个相同的区间内。如果是比对到转录本上,由于可变剪切的影响,可以适当放宽比对质量
在外显子和比对方向上的 read 覆盖率的均一性,也是评估质量的重要指标。如果 reads 主要聚集在转录本的 3' 端,可能表明原始样本的 RNA 质量较低
比对上的 reads 的 GC 含量,可能揭示了 PCR 的错误率
主要软件有:Picard、RSeQC 和 Qualimap
2.3 定量后
在计算完表达的量化值之后,可以计算 GC 含量和基因长度的误差,在必要时可以使用标准化方法来进行校正
如果参考转录组注释得很好,则可以分析样本的生物构成,来评估 RNA 纯化步骤的质量。例如,rRNA 和 small RNA 不能出现在 polyA longRNA 的制备中
NOISeq 和 EDASeq 等 R 包可以使用图形来展示 count 数据的质量控制
2.4 可重复性
上面的质量控制都只是针对单个样本的,此外,不同样本之间的可重复性评估,对于评价整个数据集的质量也是至关重要的
技术重复样本的可重现性一般很高(spearman ),但是生物学重复样本之间并没有明确的标准,取决于实验系统的异质性。如果不同实验系统之间存在差异基因,则同一条件下的生物学重复在主成分分析(
PCA)中会被聚类在一起。
3. 序列比对
在对样本的 raw reads 进行质控之后,就可以进行序列比对了,序列比对主要有三种策略,如下图
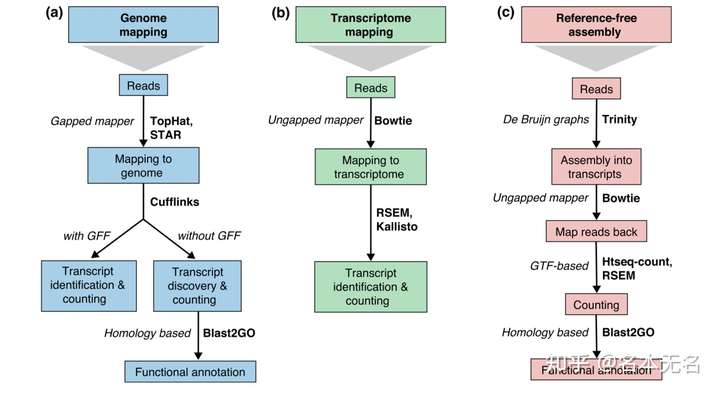 Conesa et al. Genome Biology (2016)
Conesa et al. Genome Biology (2016)
如果有参考序列,根据参考序列的不同,可以分为
- 比对到基因组:使用间隔比对算法,如
TopHat、STAR等,然后根据是否提供了注释文件(GFF格式文件,包含转录本位置信息),又可以分为转录本识别和转录本发现并进行定量分析 - 比对到转录组:使用非间隔比对算法,如
Bowtie等,然后使用RSEM或Kallisto方法识别转录本并计算定量信息
如果没有参考序列,则需要先把序列组装成转录本,再将 reads 比对到组装后的参考转录本上,然后使用 HTseq-count 等算法对转录本进行定量
3.1 转录本发现
使用 Illumina 技术检测的 short reads 来发现新的转录本是 RNA-seq 分析中的一个挑战。通常来说,短 reads 很少会跨越多个剪切位点,这就很难直接推断出一个转录本的整体长度。
此外,转录的起始和终止位置也比较难识别,一些像 GRIT 的工具,通过合并 5' 端的信息可以提高异构体识别的准确性。其他如 Cufflinks、iReckon、SLIDE 和 StringTie 等方法,通过结合现有的注释信息,作为一个可能的异构体列表
一些寻找基因的工具,如 Augustus,结合 RNA-seq 数据,可以更好的注释蛋白编码转录本,但是对非编码转录本的性能更差。
3.2 De novo 转录本重构
在没有转录本或转录本不全的情况下,可以对 reads 进行组装来重构一份转录本。可选的方法很多,如 SOAPdenovoTrans、Oases、Trans-ABySS 或 Trinity
通常来说,使用双端链特异性测序和 long reads 测序包含更多的信息,会有更好的效果
虽然,对于低表达的转录本进行组装的可靠性较低,但是 reads 太多也会导致潜在的组装错误和较长的时间消耗等问题。因此,在深度测序的样本中,可以适当减少 reads 的数量
对于多样本的比较,可以将所有样本作为一个输入来构建参考转录本,然后分别对每个样本的 reads 进行比对
无论是使用参考序列还是从头开始组装,使用短 reads 的 Illumina 技术来完全重构转录组仍然是一个具有挑战性的问题
4. 转录组定量
RNA-seq 最广泛的应用就是用来评估基因和转录本的表达,这一应用主要是基于比对到转录组区间内的 reads 的数量
最简单的方法是,使用 HTSeq-count 或 featureCounts 计算区间内的 reads 数来量化基因的表达。这种基因水平的(不是转录本水平)的量化方法使用的是 GTF 文件,这种文件包含外显子和基因在基因组上的坐标。
但一般不能直接使用 read count 来比较基因的表达水平,因为该值会受到转录本长度、reads 总数以及测序偏差等因素的影响。所以需要先进行标准化,标准化方法有
RPKM/FPKM: 每百万reads每一千碱基对中包含的reads数
该方法先计算测序深度系数,即总 reads 数除以 一百万,然后计算基因或转录本的长度(单位为 kb),标准化顺序为先消除测序深度的影响,再消除长度的影响:
其中
x表示一个基因或转录本,或基因组上一段特定的区域表示比对到
x外显子区域的reads数;R表示当前样本中包含的全部reads数x外显子区域包含的碱基数(长度,bp)
FPKM 与 RPKM 的计算公式一样,只是 RPKM 用于单端测序,FPKM 用于双端测序
TPM: 其与RPKM最大的区别是,标准化顺序为先消除基因长度的影响,再消除测序深度的影响
首先,将 reads count 除以基因或转录本的长度(kb)得到 RPK(reads per kilobase),然后将样本中所有的 RPK 加起来除以 ,得到标准化系数,最后使用
RPK 除以标注化系数
其中
x表示一个基因或转录本,或基因组上一段特定的区域x外显子区域的reads数x外显子区域包含的碱基数(kp)N表示基因或转录本总数
这样,每个样本的 TPM 总和是一样的,便于比较样本间的差异
目前,也有许多复杂的算法通过解决相关转录本共享 reads 的问题来评估转录本水平的表达,例如,Cufflinks 使用 TopHat 的比对结果,应用期望最大化算法来评估转录本的丰度。这一方法考虑到长度不同的基因的 reads 分布并不均匀等因素的影响。
还有其他算法也可以量化转录组的表达,例如 RSEM、eXpress、Sailfish 和 kallisto 等。这些方法允许转录本之间存在多比对的 reads,并输出经测序偏差校正的样本内归一化值。
5. 差异表达分析
差异表达分析是对样本间基因的表达值进行比较,虽然 RPKM、FPKM 和 TPM 标准化方法消除了测序深度和基因或转录本的长度因素的影响,但这些方法依赖于总的或有效的 reads 数,当样本的具有异质性转录本分布或当高表达或差异表达的特征扭曲了 count 分布时,表现欠佳
而像 TMM、DESeq、PoissonSeq 和 UpperQuartile 等方法会忽略高变异或高表达的特征。
干扰样本内比较的其他因素包括不同样本的转录本长度变化、转录本覆盖位置的偏差、平均片段大小以及基因的 GC 含量等
NOISeq 这个 R 包提供了多种绘图,来识别 RNA-seq 数据中的误差来源,并应用相应的方法来标准化这些误差
除了这些样本内特异的标准化方法,还需要解决数据集之间的批次效应(不同实验条件下产生的数据之间存在的差异),批次矫正方法有 COMBAT 和 ARSyN 等,虽然这些方法是针对芯片数据设计的,但是在 RNA-seq 数据中也有很好的效果
计算差异表达的方法有很多,有些方法,如 edgeR 将原始的 read counts 作为输入,并在统计模型中加入了标准化,另一些方法,需要先对数据进行标准化,如 DESeq2 使用的是负二项分布作为参考分布,并提供了自己的标准化方法。
baySeq 和 EBSeq 是贝叶斯方法,还有一些基于线性模型的方法。最后,一些非参数方法,如 NOISeq 和 SAMseq
对于小样本量的研究,负二项分布会存在噪音污染,这种情况下,一些简单点方法,如基于 Poisson 分布的 DEGseq,或者基于经验分布的 NOISeq 可能会更好些。
但是需要强调的是,在没有足够生物学重复的情况下,无法进行总体的推断,因此任何 p 值计算都是无效的。
许多独立的研究都已经证实,选择不同的方法会对结果有一定的影响,而且没有哪一种方法能够适用于所有的数据,所以,推荐在分析的时候使用多个软件进行相互验证。
6. 可变剪切分析
可变剪接(Alternative Splicing) 是指转录形成的前体 RNA 通过去除内含子、连接外显子而形成成熟 RNA 的过程,从而实现一个基因同时编码多种蛋白质,实现生物功能多样性
在不同组织或者发育的不同阶段,可变剪接不是一成不变的,在特定的组织或条件下,通过连接不同的外显子,会产生特定的剪接异构体(isoform)。有大量的研究发现,可变剪接的变化与癌症等多种疾病相关,所以研究可变剪接在不同组织中的作用是非常有意义的。
转录本水平的差异表达分析可以潜在地检测同一基因的转录异构体表达的变化,已经有一些算法应用于 RNA-seq 数据的中进行可变剪切分析
这些方法主要分为两大类:
- 异构体表达估计与差异表达相结合,来揭示总基因表达中每种异构体的比例变化
例如,BASIS 方法使用分层贝叶斯模型来直接推断转录异构体的差异表达;CuffDiff2 方法先评估异构体的表达,然后比较它们之间的差异;rSeqDiff 方法使用分层似然率检验同时检测无剪接变化的差异基因表达和差异异构体表达。
所有这些方法通常都受限于短读长测序的内在局限性,无法在异构体水平上进行准确识别
- 一种所谓的
exon-based的方法,它跳过了对异构体表达的估计,通过比较样本之间基因外显子和连接点上的reads分布来检测可变剪接的信号
其基本假设为:可以在外显子及其连接点的信号中追踪异构体表达的差异。
DEXseq 和 DSGSeq 采用类似的思路,通过检测基因的外显子(和连接点)上 read counts 的差异显著性来识别不同的异构体。
rMATS 是通过比较用连接点的 reads 定义的外显子 inclusion levels 表达水平的差异
可变剪接作为转录后修饰的重要环节,对细胞的活性和疾病的发生发展都有广泛的作用。尽管目前软件很多,但是软件间的比较却比较少。作者系统地比较了10款软件,利用了一致性(consistency)、可重复性(reproducibility)、精确度(precision)、召回率(recall)和错误发现率(false discovery rate)、报道的可变剪接基因的一致性(agreement upon reported differentially spliced genes)与功能富集分析一致性等方面来评价不同的可变剪接软件。
软件主要是三大类:
- exon-base :DEXseq、edgeR、JunctionSeq、limma
- isoform-base:cuffdiff2、diffSplice2
- event-base:dSpliceType、MAJIQ、rMATS、SUPPA
可变剪接的检测策略
isoform-base : 先构建全长转录本和定量,再来做差异分析
count-base:将基因用单个统计单元来表示,并且进行差异分析
- exon-base :利用exon来作为统计单元
- event-base :利用junction region作为统计单元
各个软件说明
isoform base
- cufflinks:首先构建overlap graphs,之后估计转录本丰度,最后检查差异基因和isoform;
- DiffSplice:基于图论的方法来进行分析,首先基于比对构建转录本,然后对不同的path来定量丰度,最终鉴定出可变剪接体。
count base
- exon-base:将序列分配到不同的特征上,例如exon或者junction;这个只能分析已知的特征,而不能推断出新的可变剪接事件;
- isoform-base:通过计算每个事件(PSI)值中的拼接百分比来量化剪接事件,PSI表示该值测量从包含该事件特定形式的基因中表达的mRNA的分数。
7. 融合分析
基因融合是指两个基因的全部或一部分的序列相互融合为一个新的基因的过程。其有可能是染色体易位、中间缺失或染色体倒置所导致的,可在 DNA 或 RNA 层面上表达。
融合基因通过基因失调、融合产生嵌合体蛋白这两种机制引发癌症的发生。
目前,RNA-seq 融合算法 100 多种,有人对常用的 15 中融合检测算法进行了比较
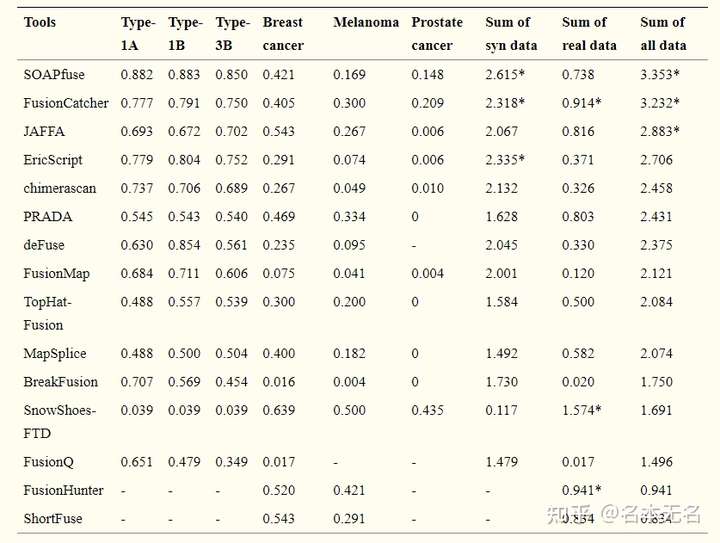 Liu et al Nucleic Acids Research, 2016
Liu et al Nucleic Acids Research, 2016
没有哪一个算法具有明显的优势,整体来看,SOAPfuse 可能会好一些,FusionCatcher 和 JAFFA 其次。
8. 功能注释
标准的转录组分析的最后一步,是使用差异表达基因来进行功能或通路的注释。最常用的两类方法是:
- 基于超几何分布的过表达富集分析
GSEA富集分析
一些工具,如 GOseq 考虑了基因长度等因素对差异表达结果的影响,并使用超几何分布进行富集分析,GSVA 和 SeqGSEA 使用类似 GSEA 的方法进行功能富集
功能富集需要预先定义的基因集合或通路,包括 GO、KEGG、Reactome 等数据库。
通过在蛋白质数据库(例如 SwissProt)和包含保守蛋白质结构域(例如 Pfam 和 InterPro)的数据库中搜索相似序列,使用直系同源分析对蛋白质编码的转录本进行功能注释。而 Rfam 数据库包含许多特征明确的 RNA 家族,例如 rRNA 或 tRNA,而 mirBase 和 Miranda 是专门研究 miRNA 的
9. 整合分析
将 RNA-seq 数据与其他类型的全基因组数据进行整合分析,使我们能够将基因表达的调控与分子生理学和功能基因组学的特定方面联系起来。
DNA测序
将 RNA 测序和 DNA 测序相结合,可以进行 SNP、RNA 编辑和表达数量性状基因座(eQTL)比对等分析。
DNA甲基化
将 DNA 甲基化和 RNA-seq 整合,可以分析差异表达基因和甲基化模式之间的相关性。使用的算法包括:广义线性模型、logistic 回归模型和经验贝叶斯模型等
- 染色体特征
通过整合 RNA-seq 和 Chip-seq 数据,可以降低 Chip-seq 分析的假阳性,并展示 TF 对其靶基因的激活或抑制作用。
MicroRNA
整合 RNA-seq 和 miRNA-seq 有可能揭示 miRNAs 对转录稳态水平的调节作用
- 蛋白质组和甲基化组
RNA-seq 与蛋白质组学的整合是有争议的,因为这两种测量结果通常显示出很低的相关性(~0.4)。尽管如此,蛋白质组学和 RNA-seq 的配对分析可用于识别新的异构体
转录组与代谢组数据的整合已被用于识别在基因表达和代谢物水平上受调控的通路,并且可以使用工具来可视化通路上下文的结果,如 MassTRIX、Paintomics、VANTED v2 和 SteinerNet
- END -
更新:更详细了一点:https://www.jianshu.com/p/2055db183907

 浙公网安备 33010602011771号
浙公网安备 33010602011771号