
读文献-Common variants conferring risk of schizophrenia A pathway analysis of GWAS data
Profiles
文章题目: Common variants conferring risk of schizophrenia: A pathway analysis of GWAS data
不想看英文题目: 精神分裂症风险的常见变异:GWAS数据的路径分析
杂志和影响因子: SCHIZOPHRENIA RESEARCH(IF: 4.939; Q1)
研究意义:
摘要: 与全基因组关联研究(GWAS)中典型的单标记分析不同,我们将基因集富集分析(GSEA)和超几何检验结合起来,利用Fisher的合并p值方法进行基于通路的分析,检测基因对介导精神分裂症的联合作用.
结论:
- 从GAIN数据集中得到GWAS数据,作者在通路水平而不是SNP水平去确定精神分裂的遗传联系.发现这些通路中涉及的基因未被单标记分析检测到.在重复研究中,这些基因的确认将保证在复杂疾病的研究中更广泛地应用基于通路的方法.
阅读中遇到的主要障碍: 无
个人看法和体会: 优点是引入了一个新的组合方式和思路.通过找到
各类注释意义:
| 注释类型 | 注释意义 |
|---|---|
| 红色 | 完全不懂的知识点,比如第一次接触的公式或算法等等(快捷键:tjys添加颜色的拼音首字母) |
| 高亮 | 一些重要知识点或者以后可以借鉴的方法(快捷键:Ctrl+H) |
| 加粗字体 | (快捷键:Ctrl+B) |
Introduction
作者为了减小偏差,使用两种统计方法去确定过表达的通路.第一时候GSEA,第二是超几何分布检验.然后再把p值合并.确定了4个通路p是小于0.05
Method and Analysis
GWAS data preparation
GWAS数据获得和质控:
我们排除了缺失基因型率为大于0.1的snp,次要等位基因频率(MAF)小于0.01或哈迪温伯格平衡(HWE)≤0.001.结果共产生72.5万个SNPs。根据之前的分析(指的是这个公共数据集发布的时候的分析),在欧洲血统的GAIN样本中没有发现明显的分层.
我们使用基本等位基因测试(卡方,1df)来计算每个SNP与精神分裂症的关联.所有的p值都经过膨胀系数λ进行校正.
如果它位于该基因内,或者位于该基因的上游或下游20kb处,我们将SNP定位到一个基因上。在后续的分析中,选择该基因中最显著的SNP来代表该基因的关联.
从MSigDB下载了通路数据,其包括了KEGG和BioCarta的通路.为了避免随机偏差或测试过于普遍的生物过程,我们丢弃了包含少于10个或超过250个基因的路径.
经过snp-基因和基因-通路的定位,我们得到了399808个snp,共定位到19896个蛋白质编码基因,涉及511条生物通路.
Gene Set Enrichment Analysis (GSEA)
总结他的Method和Analysis
-
它先得到SNP-Gene-Pathway的对应关系,这是GWAS数据预处理.
-
后面计算GSEA:
-
GSEA的第一步与常规的不一样,是计算每一个SNP的 \(χ^2\)值,选择最大的 \(χ^2\)表示这个基因与疾病的关联程度.这就把SNP的数据转移到Gene上面了.之后在对刚刚得到的基因数据进行排序,使相关性较强的基因排在列表顶端
-
接下来事GSEA的常规流程,对于每一个通路都去计算它的ES值根据一下公式:
\(ES(S) = {\mathop \max}_{1 \leq i\leq N} \begin{Bmatrix} \sum_{g_j\in S,j\leq i} \frac{{|r_{j}|}^{m}}{N_R}-\sum_{g_j\notin S,j\leq i}\frac{1}{N-N_H} \end{Bmatrix}\)
N为GWAS研究中包含的基因总数,i为基因表N中的位置,j为基因表N中在i之前的位置,\(r_j\)为基因j的χ2统计值,g代表一个基因,\(N_H\)是感兴趣途径中的基因数量.值得注意的是,当m = 0时,ES(S)简化为Kolmogorov-Smirnov检验。我们设m= 1,就像在最初的GSEA应用程序中使用的那样,通过它们的关联水平(\(r_j\))来衡量基因.ES测量路径偏离随机行走的最大偏差.
-
通过交换病例与对照的标签,对原始的GWAS进行permutation,虽然是随机的,但是会保持相同的病例/对照的比例.这一步的目的是检测富集的通路是否也与疾病显著相关,并使不同通路的ES(S)具有可比性.我们进行了10000次排列。对于每一个排列(π),我们计算ES(S)并将permutation生成的值表示为ES(S,π)。然后,对每条路径,根据10000 ES(S,π)对原始ES(S)进行归一化:
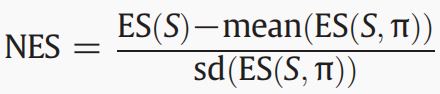
这样,对于每个通路,ES(S)和ES(S,π)在相同的背景分布下,从通路大小、基因长度、SNP密度等方面进行比较.具体来说,这种方法有效地避免了大脑或神经相关基因的基因长度偏差,这些基因往往很大。在归一化过程中,ES(S)和ES(S,π)的比较基于同一基因集;因此,对基因长度或SNP密度没有偏见.所得到的NES(S)呈正态分布,且具有可比性,不存在偏倚,尤其对长基因和SNPs数量密集的通路.通过计算ES(S,π)大于或等于实际情况的排列数,然后除以总排列数,计算出每个路径的nominal P.
-
-
计算超几何分布:
-
为了检验一个基因集在GWAS数据集中是否被超几何分布过度表示,我们首先定义了“有趣基因”。如果标记到该基因的增益标记为P<0.01,则选择该基因为感兴趣基因.这里的marker gene应该是指做完等位基因关联分析后p<0.01.这个p值阈值是任意的,但作为第一步似乎是有用的。
-
然后开始假设,假设1,L是一个基因组中考虑的基因总数(即以GWAS数据表示,并有pathway注释),M为L和中有趣基因的数量.
-
假设2,对于一个基因集(即一条路径),S是其中的基因数量L和x为M内的基因数,基于超几何分布的P值可计算为:
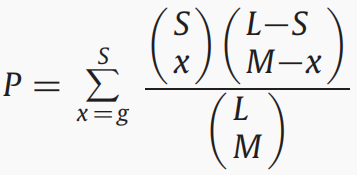
-
这个P值表示在当前基因集中观察到至少g个基因的概率。与GSEA相似,我们进行了排列,估计了nominal P值.使用Benjamini进行多次测试校正.
-
-
Fisher’s Method
-
将来自不同检验的多个P值合并,\(P_i\)是第i个测试的P值,k是测试的总数.\(\chi^2\)是一个有2k个自由度的卡方分布。我们使用Fisher的方法将每种方法计算的每种途径的nominal P值结合起来,以确定两种方法显示一致意义的途径。
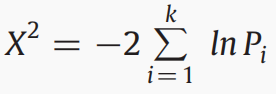
-

 浙公网安备 33010602011771号
浙公网安备 33010602011771号