
读文献--Inferring Pathway Activity toward Precise Disease Classification
Inferring Pathway Activity toward Precise Disease Classification文献解读
Introduction
Gene标记物的选择是通过对每个个体基因的表达模式在区分不同类型疾病或病例与对照组之间的能力进行评分。新患者的疾病状态是使用调整到标记基因表达水平的分类器来预测的。然而基于表达的分类面临的一个挑战是,在复杂疾病中,组织内的细胞异质性和患者之间的遗传异质性可能会削弱个体基因的辨别能力.并且通过这个方式挑选出来的Gene是与蛋白复合物或信号通路独立的(也就是不通过这些东西去挑选,单单通过表达量去挑选).这样会有冗余数据出现,影响分类器的性能.
因为这些问题,所以很多实验室开始使用更有效的标记识别方法,即将属于共同pathway的基因组的基因表达测量结合在一起.这些通路来自KEGG和GO等数据库.并且最近开始将这些基于通路的分析用于疾病诊断中去了.
一些方法使用参数化的基因表达,通过概括成员基因表达值的功能来表示通路活性.另一些方法则基于基因表达变化的一致性来估计通路激活的概率.
这些方法已经证明了与传统的基于基因的分类器相比的分类准确性,同时对于为什么表达谱与特定类型的疾病相关这一问题提供了强有力的生物学解释.
另一方面,当前基于pathway的分类器的一个潜在缺点是,预先定义的组成路径的一组基因可能来自与疾病无关的条件.此外,并不是所有的成员基因在一个被干扰的途径是典型的改变在mRNA水平.
在这里,我们提出了一种新的基于基因表达的诊断方法,以一种条件特异性的方式整合pathway信息(Pathway Activity inference using Condition-responsive genes,PAC).这些标记不是作为单个基因编码的,也不是作为文献整理的静态pathway,而是作为条件响应共同功能基因(Condition-Responsive Genes,CORGs). 为了最佳的区分不同表型的样本,我们在特定疾病的背景下从每个静态pathway中识别CORGs.将CORGs的联合表达水平视为通路“活性”,用于构建预测新患者疾病状态的分类器.
---注: 这个CORGs是作者最早提出的名词
基于路径的方法,作者在跨7个不同的数据集分类样本方面上面测试,发现优于先前的差异表达分析方法。与使用各种统计方法去推断参与共同路径的所有基因的汇总pathway activity相比,使用CORGs推断的pathway activity有更好的分类性能。
Methods
Datasets
24个被肿瘤坏死因子刺激后HeLa细胞的表达谱,62例原发性前列腺癌和41例正常前列腺癌的表达谱,143例急性淋巴细胞白血病(ALL)患者的表达谱分析,295例荷兰乳腺癌患者的乳腺癌表达谱和286例美国乳腺癌患者的乳腺癌表达谱,86例来自密歇根的患者的肺癌表达谱和来自波士顿的62名病人.
肿瘤坏死因子(TNF)研究,12个样本有正常的IkB蛋白(标记“野生型”)和12个样本表达突变IkB阻断NFkB信号(标记为“突变型”)用于前列腺癌研究,62例来自原发肿瘤(癌症),41例为正常前列腺标本(正常)白血病中,79为TEL-AML1.其他63为超二倍体HH乳腺癌数据中把转移和为转移分为两组
肺癌中,根据预后进行分型,差是一组,良好是一组.
对于patway信息,我们使用从MsigDB v1.0下载的C2功能集.包括472个典型的代谢和信号通路,汇集了8个人工管理的数据库,以及从各种微阵列研究中获得的50个共同表达的基因簇。每一条通路或基因簇都定义了一组基因(基因簇从此也被称为“pathway”)。总的来说,所涵盖的可用途径由于使用了不同的阵列平台,在7个基因表达数据集中测量了5602个基因的其中大部分(但不是全部)
Condition-Responsive Gene Identification and Pathway Activity Inference
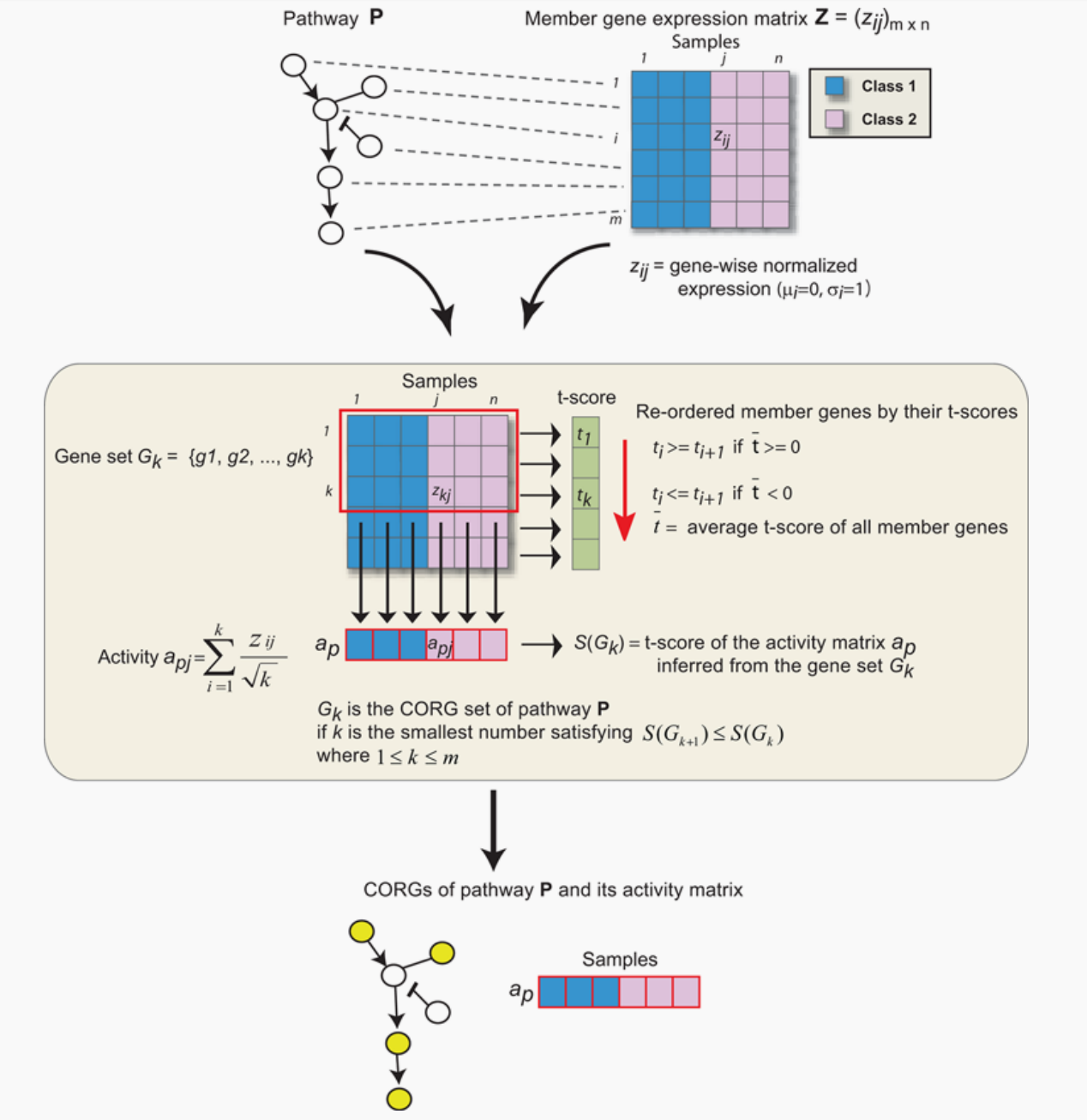
为了整合表达和通路数据集,我们将每个基因在每个通路中对应蛋白上的表达值叠加。在每个途径中,我们搜索成员基因的子集,其组合表达水平跨样本是高度区分的表型感兴趣(图1)。对于特定的基因集合G,让a表示其对研究样本的活动得分向量,让c表示相应的类别标签向量(如预后好与坏)。为了得到a,表达式值gij被归一化为z转换得分zij,在所有样本j中,每个基因i的均值mi= 0,标准差si= 1。将基因集合中每个成员基因的单个zij求均值,得到一个组合z分数,即活动指数aj(分母中使用成员基因数的平方根来稳定均值的方差)。许多类型的统计,如Wilcoxon score或Pearson correlation,可以用来对a和c之间的关系进行评分。在本研究中,我们将判别得分S(G)定义为c定义的组样本之间对a推导出的t检验统计量[32]。
对于给定的路径,采用贪心搜索来确定路径中S(G)局部最大的成员基因子集。我们把这个子集称为“条件响应基因”(CORGs)集合,代表了相关条件下的大部分通路激活。识别在CORG集中,成员基因的t检验分数首先排序,如果所有成员基因的平均t检验分数为负,则由高到低排序,反之则由低到低排序。CORG集将G初始化为只包含顶级成员基因,并迭代扩展。在每次迭代中,考虑添加t检验分数次优的基因,当不添加增加判别分数时,搜索终止.将最终CORG集的活性向量a视为跨样本的通路活性。
Previous Gene-Set Ranking Approaches and Other Pathway-Based Classification Methods
Tian et al.[16]提出的方法,根据其所有成员基因的表达与疾病表型之间的相关性,来评估通路在疾病中被改变的概率.对于MsigDB中的每个通路P, 通过将所有成员基因的T检验统计分数取平均数,计算出得分T。T值越高,表明该通路与疾病状态的相关性越强。每个数据集的前10%路径(52条路径)被选择用于进一步分析和分类。决定一条通路是否被疾病破坏的依据是成员基因在兴趣类之间的鉴别能力(使用t检验统计量).然而,可能存在一些路径,它们的中断的特征与当前的分类任务是无关的(独立)。为了检测这样的信号,可以在Tian等人的框架内采用若干统计函数。与t检验不同,这些函数的设计目的是检测受干扰的模式,而不是平均表达变化。
为了比较作者的PAC方法与其他activity推断方案,我们实施了另外三种表达式摘要方法:包括主成分分析(PCA),均值和中值方法. Bild等人用成员基因表达的第一个主成分来表示给定通路的激活,而Guo等人用均值和中位数等简单统计来总结成员基因的表达水平。
Marker Robustness Evaluation
对于每个数据集,对数据集中的每个mRNA表达谱生成100个可选的两次分割.使用Tian等人的方法
这一部分看不懂什么是two-fold splits
Classification Evaluation
Logistic回归模型e在通路活性矩阵(通路vs样本)和原始基因表达矩阵(基因vs样本——即。传统的基于基因的分类)中被训练的。在数据集内实验中,将数据集中的表达样本进行分割,其中五分之四的样本作为训练集构建分类器,五分之一的样本作为测试集(五倍交叉验证)。在标记选择(包括CORG识别)和分类器训练期间,数据集中的五个子集依次作为测试集进行评估和保留.为了训练一个广义分类器,并最小化过拟合,我们进一步将训练集分成三个较小的等大小子集:两个子集被用作标记选择集,以对标记(通路或基因)进行排序,并识别corg(仅通路),一个子集被用作验证集,以评估哪些标记集对分类有意义。因此,根据标记选择集中使用的样本,特定路径的corg可能不同。根据判别能力的p值对路径或基因进行排序,对标记选择集中的样本进行分类,然后按照p值的递增顺序依次添加标记,建立logistic回归模型。通过评估ROC曲线下面积(Area Under ROC Curve,见AUC)来优化分类器中使用的验证集的标记物数量.AUC度量捕获了整个灵敏度/特异性值范围内的性能。最终的分类性能被报告为使用验证集优化的分类器在测试集上的AUC。为了进行无偏评估,我们在每个数据集中生成了100个可选的5倍分割样本,并对每个分割进行交叉验证。最终报告的AUC值在500种随机选择的方法中平均,将数据划分为五分之四的训练样本和五分之一的测试样本.为了进行无偏评估,我们在每个数据集中生成了100个可选的5倍分割样本,并对每个分割进行交叉验证。最终报告的AUC值在500种随机选择的方法中平均,将数据划分为五分之四的训练样本和五分之一的测试样本。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号