scRNAseq benchmark 学习笔记
 A comparison of automatic cell identification methods for single-cell RNA sequencing data
A comparison of automatic cell identification methods for single-cell RNA sequencing data
背景
把早年没填完的坑(单细胞测序的细胞类型鉴别)给重新拾起来
其Github描述的基本情况:
- 作者并不对单个分类器进行说明,统一包装在benchmark工程里,还建立了docker容器
- 但说明了在scripts里有封装好的方法,可以直接读入数据进行分类
- 可以使用,但是整个过程需要知道,读全文
回顾文章
1. 文章采用的方法和数据集介绍
- 22个方法,27个数据集(前十一个用于数据集内比较不同方法,PBMC用于数据集间)
- 有在单个数据集里评估各个分类器的实验,也有在不同数据集评估单个分类器的实验
- 方法有两类:一是需要标记好细胞群体种类(即已分类的数据集)来训练的监督式方法,二是需要可以分辨细胞群体种类的mark基因作为输入的先验知识方法和针对特殊细胞群体种类已预训练的特殊分类器的先验知识方法
- 数据集涵盖不同细胞数目的、不同数目的基因的、不同细胞群体种类数目的、还包含可以用于先验知识方法的数据集
- 数据集主要有:胰腺数据集(包含不同细胞数目的、人和小鼠的、不同测序技术的)、小鼠大脑数据集(对细胞需要有不同注释深度的,即分类目标是分几类,分类目标群体数越多就分得越细,分类目标群体数越多容易让分类器分不出这个细胞究竟属于哪类)、大数据集(Tabula Muris (TM) and Zheng 68K,细胞数目多同时细胞群体也多)、对细胞群体已排序的数据集(没找到其特征)
2. 结果
前置知识
- 先验知识方法必须要有相应mark基因(可以是训练集中已知的差异基因)或者已预训练的分类器,所以没法对所有数据集进行分类,即这个评估大部分数据集只有监督式方法的,PMBC的两个数据集可以评估所有方法
- 先验知识方法如果是需要mark基因的,还探究了不同数目mark基因的效果
- 如果光用median F1-score评价的话,有失偏颇,因为有的分类器带有拒绝选项即对不够自信的某次分类进行拒绝,对细胞打上“未标记”标签,这样会避免盲目分类拉低median F1-score
- 定义数据集复杂性:获得每个细胞群体的全部基因表达量比如记为A,计算各个群体之间A的相关性,取各个群体的最大相关累加取平均值记为复杂性
单个数据集中各个方法的评估
测试场景:对于监督式:用已知细胞群体分类的数据集(要求数据集有已分类的数据集)来训练,再对数据集进行分类;对于先验知识类型:具备mark基因或者预训练的前提。
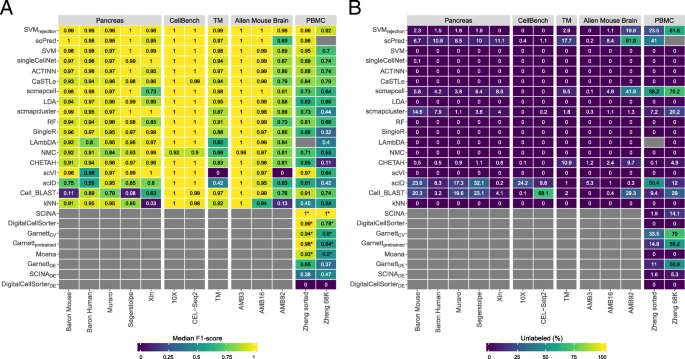
图注:DE下标是依赖于mark基因(此处是差异基因)的先验知识方法
小结:
- 从胰腺数据集来看,SVM和SVMrejction总体优于其他监督式分类器
- 从小鼠大脑数据来看,在不同注释深度的实验中,一般目的分类器(SVMrejection, SVM, and LDA)优于单细胞特异性目的的分类器
- 先验知识方法表现并没有在单个数据集中有所提高
- 大数据集且注释深度较高的,SVMrejection拒绝率较高,较好的是SCINA,但是需要先验知识(mark基因或预训练)
- 先验知识分类器依赖于mark基因的数目和特异性(方法原作者挑选还是基于差异基因),所以选择mark基因的数目和决定哪些作为mark基因是个艰巨任务
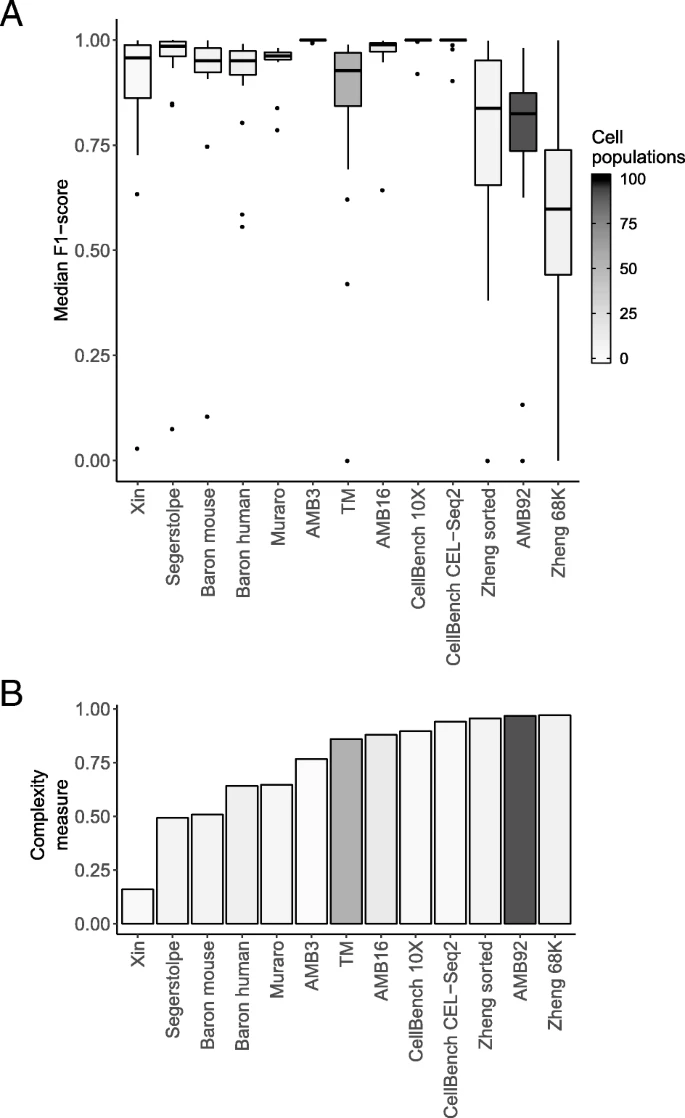
- 复杂性和细胞群体数目影响median F1-score,且似乎前者影响更大
数据集间单个方法的评估
测试场景:总体是用一个数据集训练用另一个数据集来评估,先测试只有测序方式差别的两个数据集;其次测试PBMC1和2共42种组合(PBMC1有7种测序方式,PBMC2有6种),一个用于训练一个用于分类;还有两个数据集采用同一测序方式的数据集的评估。
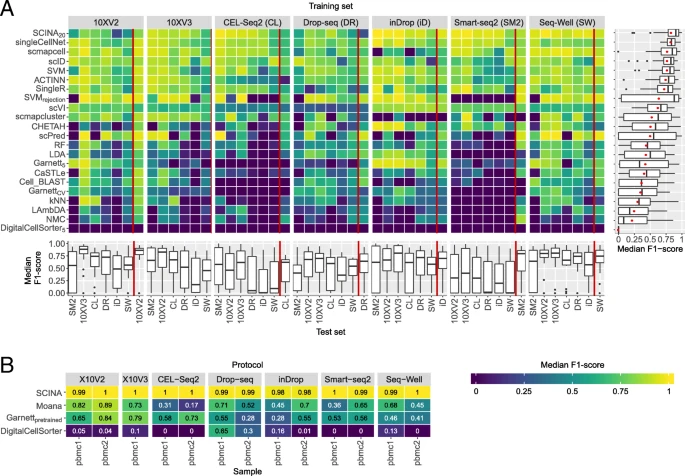
- 红线以左代表以灰色块的测序方式训练,以PBMC1其他测序方式来评估的结果
- 红线以右代表以灰色PBMC1来训练,PBMC2来评估
- 右边箱线图是总体表现
- 下方是先验知识类型的分类器的表现,有两种样本的不同测序方式的评估
跨越物种、单细胞测序与单神经元测序、不同注释深度数据集
测试场景:只对于监督式,列标签用于训练,灰色块用于分类,在一个数据集训练在灰色块进行分类
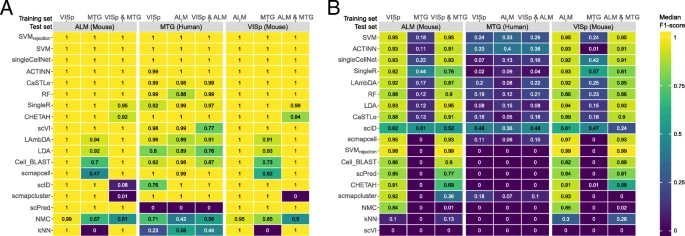
- 深度注释的数据集分类很难
用MNN法合并三个数据集用于训练,另一数据集用于分类
测试场景:合并或者不合并
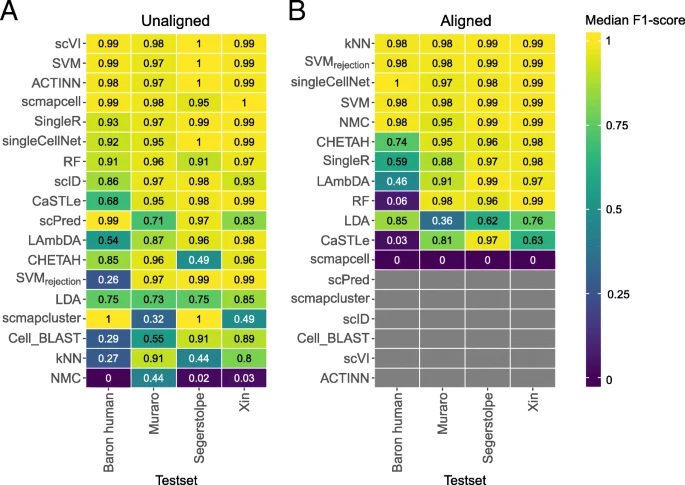
- 合并后SVMrejection有提高,总体SVM表现佳
拒绝选项评估
前置知识
- 拒绝选项是用来标记在训练集中没有的细胞
- 最好的用法是在某组织的细胞数据集训练,就在某组织细胞的数据集分类
- 拒绝选项的优劣是,如果是未知细胞就应该全数拒绝就是好的
- 图B是仅仅排除某个细胞种类,且用此细胞种类的数据来训练
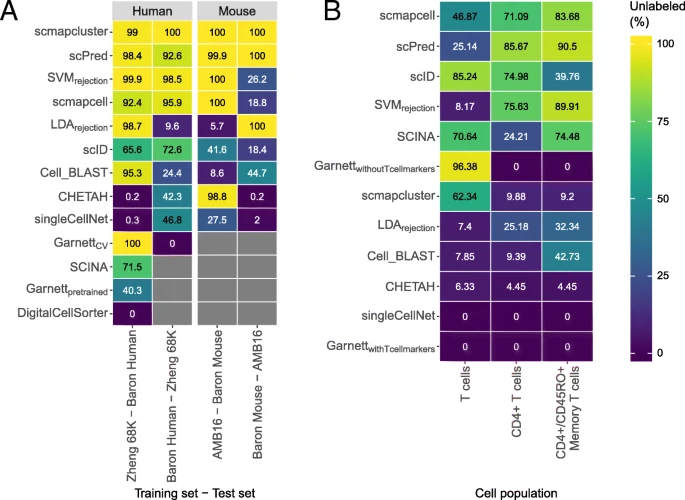
基因输入特点的评估
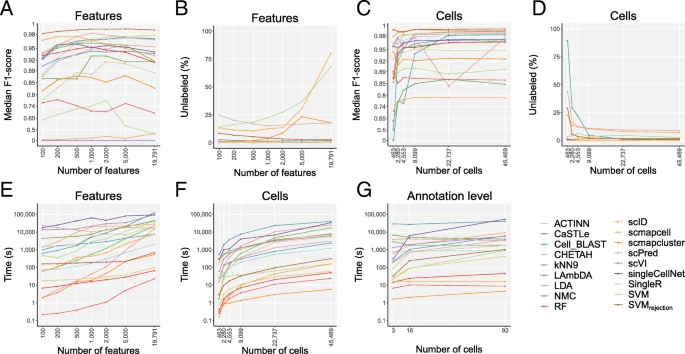
图注参考原文
- 一些分类方法会存在过训练的现象,导致效能降低
- 总体SVM和SVMrejection效能较好
- 细胞数目影响不大,分类数据集增大不一定带来runtime增加
- scmapcell, scmapcluster, SVM, RF, and NMC runtime都在6min以下
3. 讨论
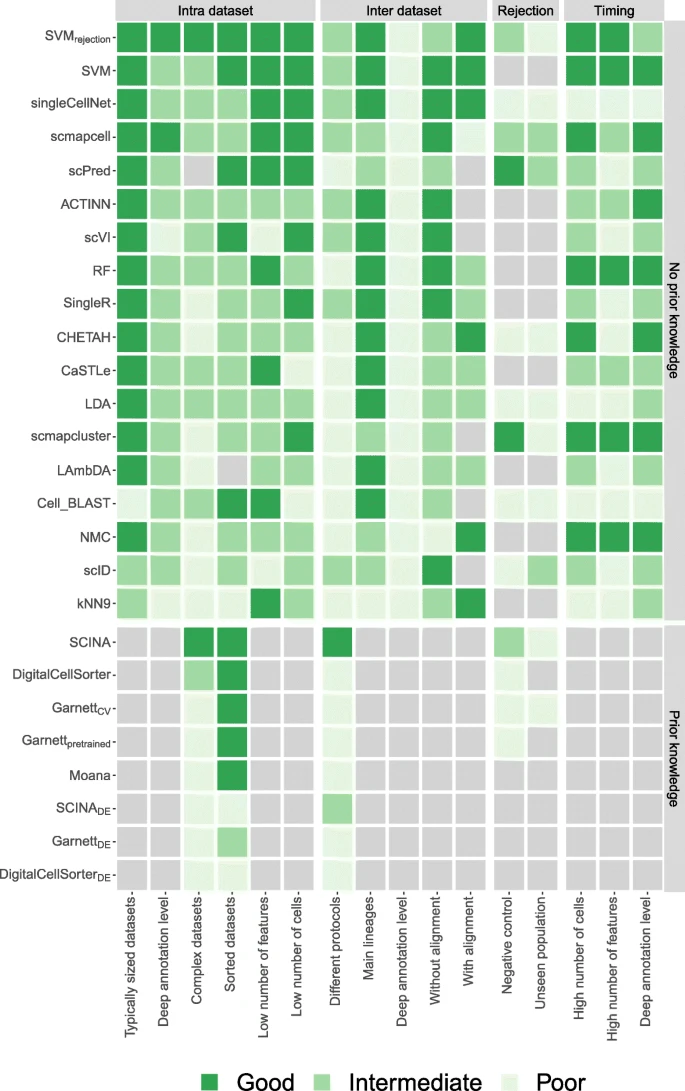
总结
4.方法
值得注意的点:
- SVMrejection的拒绝阈值是可以设置,本文设置为0.7
- 大多数方法都有内置的标准化,即log转换
数据处理:
- 基于人工注释结果去除双峰、碎片、未标记细胞
- 去除在所有数据集所有细胞中count为0的基因
- 计算所有细胞可探测到基因的数目,再计算所有细胞探测到基因数目的中位数,筛除低于3倍MAD的细胞
- 交叉验证之前剔除了小于10个细胞群体的数据集
- mark基因的评估基于四个特点(1) the number of marker genes, (2) the mean expression, (3) the average dropout rate, and (4) the average beta of the marker genes [37].
- 选择差异表达基因作为mark基因的操作是基于CPM标准化加上log转换,MAST作为one-vs-all的差异基因寻找工具,差异基因只取正数的FC
- 数据集之间探究基因特征选取时只用了所有数据集共有基因
- 不同测序技术数据集之间的评估中,用MNN法修正技术误差和批次效应,交叉验证采用的数据集单元作为fold,即某个数据集作为分类数据集,其他的三个为训练数据集
- 评估矩阵即评估的指标:1、F1分值中位数;2、unlabel rate、3、计算时间
实战
本文仅仅针对某个方法进行使用,故不全然使用其benchmark工程。参考其Github
- 先下载数据
- 安装R3.6和python3.7,版本参考文章当时的最新版本,并安装对应R包和python模块
- 参考Github的General Usage,注意用法:
$ Rscripts Cross_Validation.R "CEL-Seq/CL_pbmc1Labels.csv" 1 "CEL-Seq"
$ python
>>> from run_SVMrejection import run_SVM
>>> run_SVM("CEL-Seq/CL_pbmc1.csv","CEL-Seq/CL_pbmc1Labels.csv","CEL-Seq/CV_folds.RData","Results/CEL-Seq/pbmc1")
$ R
> result <- evaluate('./Results/CEL-Seq/pbmc1/SVM_True_Labels.csv', './Results/CEL-Seq/pbmc1/SVM_Pred_Labels.csv') # evaluate.R同级目录下
> capture.output(result, file = "Results/CEL-Seq/pbmc1/SVM_evaluation.result")" # 保存list的方法
结果包含 the corresponding accuracy, median F1-score, F1-scores for all cell populations, % unlabeled cells, and confusion matrix. 混淆矩阵是衡量细胞群体相似性的
遇到的问题
- unzip: cannot find zipfile directory in one of... 是压缩文件大小超过2GB,不能使用默认的unzip,用7z

