

首先,所有的以及人特异性的序列的转录因子的结果都已经出来了,这次想要按照重复家族,来试图解释:
(1)我们已经有人表达特异性的基因和转录因子的数据集(师姐之前给过我)
(2)不同的重复序列家族,结合的转录因子是否相同(从整体上看,也许也有一些文献的证据)
(3)有否存在特异性的和插入序列家族结合的转录因子(如果有,有什么功能,是否也是人特异性的)
一、提取不同家族的比对的结果
刚刚看了一下,也许是需要重新的比对,因为它odds计算的时候,是从整体上进行计算的。所以,还需要继续拆分reads。
cp all_repeat.bed ./sub_family/
cp ./new_trial/human_specific_repeat_uniq.bed ./sub_family/
cd sub_family/
grep 'SVA' all_repeat.bed >./all_SVA.bed
grep 'SVA' human_specific_repeat_uniq.bed >./Hs_SVA.bed
mv Hs_SVA.bed ./SVA
mv all_SVA.bed ./SVA
grep 'SINE' all_repeat.bed >./SINE/all_SINE.bed
grep 'SINE' human_specific_repeat_uniq.bed >./SINE/Hs_SINE.bed
grep 'LINE' all_repeat.bed >./LINE/all_LINE.bed
grep 'LINE' human_specific_repeat_uniq.bed >./LINE/Hs_LINE.bed
grep 'LTR' all_repeat.bed >./LTR/all_LTR.bed
grep 'LTR' human_specific_repeat_uniq.bed >./LTR/Hs_LTR.bed
bgzip -c Hs_SINE.bed >Hs_SINE.bed.gz
time giggle search -i /home/xxzhang/workplace/project/giggle_workplace/human_factor_index -q Hs_SINE.bed.gz -s > Hs_SINE.result
bgzip -c all_SINE.bed >all_SINE.bed.gz
time giggle search -i /home/xxzhang/workplace/project/giggle_workplace/human_factor_index -q all_SINE.bed.gz -s > all_SINE.result
其余的几个家族的序列全部按照这种情况进行分析比较。
二、odds_ratio到底指的是什么?
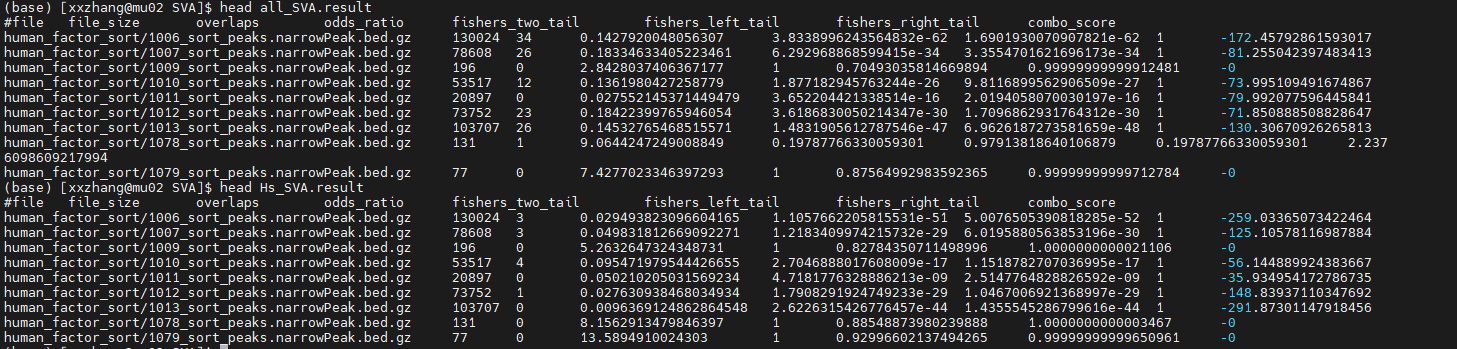
我们发现:overlap为0的转录因子,其odds_ratio竟然特别的大(按理说,ratio越大,表示的是这个转录因子的peak越富集在我们候选的区间中)。这样的结果却是让我充满怀疑odds_ratio适不适合作为判断富集程度的指标?
【注】我觉得对于底层原理的不理解,总是让我和“真相”隔着一层暧昧的纱,心里很不爽快。既然自己有时间有精力,为什么不用心的把真相挖出来,我倒要看看怎么处理更加的合适?
odds ratio是统计学上的基本概念,我们将其称之为“优势比”。==> 后来发现,用作者定义的combine score来评估转录因子的特性,似乎更符合生物学的情况。
通过对原始文献的阅读,发现自己有很多可以做的事情,还有之前的自己“理解错的”地方,这大概就是学习的过程。
(二)combine_score的再处理
使用R对所有的repeat 序列以及人特异性的repeat 序列分别比较所得到的comb_score进行比较,绘制散点图,也趁机把代码整理了一下。得到了一些pattern,但是是否真实未知。
awk '{print $1,$8}' all_SINE.result |sed 's/human_factor_sort\///g' |sed 's/_sort_peaks.narrowPeak.bed.gz//g' |sed 's/#file/DCid/g' |sed 's/combo_score/all_combo_score/g' >all_SINE.txt
awk '{print $1,$8}' Hs_SINE.result |sed 's/human_factor_sort\///g' |sed 's/_sort_peaks.narrowPeak.bed.gz//g' |sed 's/#file/DCid/g' |sed 's/combo_score/Hs_combo_score/g' >Hs_SINE.txt
我突然觉得从某种意义上将,比较 人特异性的重复序列 和 非人特异性的重复序列是不是更加有生物学意义。因为所有的人特异性的重复序列中包含了人特异性的这块,所以我不知道这是否会影响最终的结果。
现在尝试找到,在all中有,而在hs文件中没有的序列,重新进行分析。
(base) [xxzhang@mu02 all_not_Hs]$ awk '{print $1,$2,$3,$6}' all_SINE.bed >all_SINE_2.bed
(base) [xxzhang@mu02 all_not_Hs]$ awk '{print $1,$2,$3,$6}' Hs_SINE.bed >Hs_SINE_2.bed
grep -vFf Hs_SVA_2.bed all_SVA_2.bed >all_not_Hs_SVA.txt #用grep实现了功能。
sed 's/\s\+/\t/g' all_not_Hs_SVA.txt >all_not_Hs_SVA.bed #替换空格
vim all_not_Hs_SVA.bed
rm all_not_Hs_SVA.txt
rm all_SVA*
rm H* #删除多余的文件
bgzip -c all_not_Hs_SVA.bed >all_not_Hs_SVA.bed.gz
time giggle search -i /home/xxzhang/workplace/project/giggle_workplace/human_factor_index -q all_not_Hs_SVA.bed.gz -s >all_not_Hs_SVA.result
awk '{print $1,$8}' all_not_Hs_SVA.result |sed 's/human_factor_sort\///g' |sed 's/_sort_peaks.narrowPeak.bed.gz//g' |sed 's/#file/DCid/g' |sed 's/combo_score/all_not_Hs_combo_score/g' >all_not_Hs_SVA.txt
#接下来的过程就很相似了,不知道找到的东西的结果对不对?#反正先把他做出来,然后再一起来看
#其他的重复序列,过程类似。
处理了,但是结果并不是特别的理想,找到了一些转录因子,但是这个转录因子的特征在不同的细胞类型之间变异很大,让人一时之间有点不知道怎么办才好。
(三)处理染色质开放的数据(acc)
1、解压提取peak文件
(base) [xxzhang@mu02 human_chromAcc]$ tar -xf R56Q7GGRZEY7L4PH4RA9.tar
(base) [xxzhang@mu02 human_chromAcc]$ gunzip human_ca.tar.gz
(base) [xxzhang@mu02 human_chromAcc]$ tar -xf human_ca.tar
所有的染色质开放的文件都保存在了human_ca文件夹下。
现在同样适用giggle,对其构建索引,进行进一步的处理。
目前这些数据存放在目录文件夹下:/home/xxzhang/data/Epigenome/cistrome/human_chromAcc/human_ca/
/home/xxzhang/workplace/software/giggle/scripts/sort_bed "/home/xxzhang/data/Epigenome/cistrome/human_chromAcc/human_ca/*.bed" human_ca_sort/ 32
giggle index -i "human_ca_sort/*.gz" -o human_ca_index -f -s
(base) [xxzhang@mu02 giggle_workplace]$ cd /home/xxzhang/workplace/project/giggle_workplace/sub_family/LINE/
(base) [xxzhang@mu02 LINE]$ time giggle search -i /home/xxzhang/workplace/project/giggle_workplace/human_ca_index -q Hs_LINE.bed.gz -s >Hs_LINE_ca.result
real 0m2.093s
user 0m1.178s
sys 0m0.231s
(base) [xxzhang@mu02 LINE]$ awk '{print $1,$8}' Hs_LINE_ca.result |sed 's/human_ca_sort\///g' |sed 's/_sort_peaks.narrowPeak.bed.gz//g' |sed 's/#file/DCid/g' |sed 's/combo_score/Hs_combo_score/g' >Hs_LINE_ca.txt
(base) [xxzhang@mu02 all_not_Hs]$ awk '{print $1,$2,$3,$6}' all_LINE.bed >all_LINE_v2.bed
(base) [xxzhang@mu02 all_not_Hs]$ awk '{print $1,$2,$3,$6}' Hs_LINE.bed >Hs_LINE_v2.bed
(base) [xxzhang@mu02 all_not_Hs]$ grep -vFf Hs_LINE_v2.bed all_LINE_v2.bed |sed 's/\s\+/\t/g' >all_not_Hs_LINE.bed
(base) [xxzhang@mu02 all_not_Hs]$ bzip -c all_not_Hs_LINE.bed >all_not_Hs_LINE.bed.gz
bash: bzip: command not found...
Similar command is: 'zip'
(base) [xxzhang@mu02 all_not_Hs]$ bgzip -c all_not_Hs_LINE.bed >all_not_Hs_LINE.bed.gz
(base) [xxzhang@mu02 all_not_Hs]$ time giggle search -i /home/xxzhang/workplace/project/giggle_workplace/human_ca_index -q all_not_Hs_LINE.bed.gz -s >all_not_Hs_LINE_ca.result
awk '{print $1,$8}' Hs_LINE_ca.result |sed 's/human_ca_sort\///g' |sed 's/_sort_peaks.narrowPeak.bed.gz//g' |sed 's/#file/DCid/g' |sed 's/combo_score/Hs_combo_score/g' >Hs_LINE_ca.txt
现在似乎卡住了,想要得到具体的某一个候选的片段,有哪些reads比对上,结果报错:
(base) [xxzhang@mu02 SVA]$ time giggle search -i /home/xxzhang/workplace/project/giggle_workplace/human_ca_index -q Hs_SVA.bed.gz -v -o |head
Could not open file 'human_ca_sort/79702_sort_peaks.narrowPeak.bed.gz'
giggle: Could not open human_ca_sort/79702_sort_peaks.narrowPeak.bed.gz.
##chr10 100092148 100093575 SVA_F SVA SVA
说无法打开某文件,我现在也依旧比较困扰,该如何解决它。
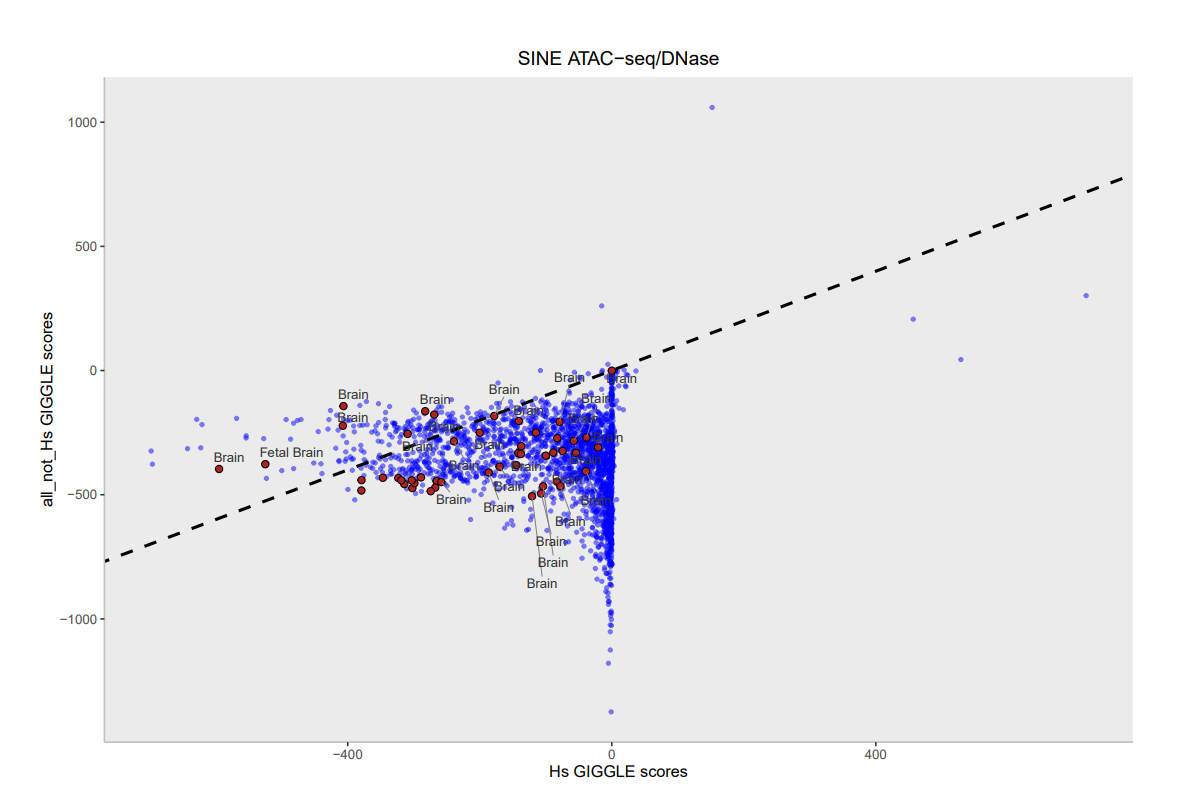
而通过对GIGGLE score的查找,我们发现,brain的情况依旧比较糟糕,且也仅有部分(个位数)的样本的数据集是在我们认定的“显著”的范围内的。
我不知道为什么,在转录因子的数据中也是同样的结果。难道这些序列从整体上看,富集的并不显著嘛?还是我们不能如此武断的解释,我们需要结合其他方面的一些证据,来说明这个问题。
是(1)技术的问题:call peak的时候,把重复序列筛掉了?我们这种方法,这个giggle score的值根本就不能说明你想要的探究的问题?
(2)生物学的问题:确实是这样的一种情况?
(三)处理组蛋白修饰的数据
/home/xxzhang/data/Epigenome/cistrome/human_histone_mark/human_hm/
这里还可以做一下分组处理,一个二个boxplot的分组的图。
建索引的过程是相似的。
1、建索引。
#首先对原始的bed文件,进行压缩和排序。使用giggle内置的脚本来实现。
mkdir human_hm_sort
/home/xxzhang/workplace/software/giggle/scripts/sort_bed "/home/xxzhang/data/Epigenome/cistrome/human_histone_mark/human_hm/*.bed" human_hm_sort/ 32
#构建名为human_hm_index的索引,这个时间稍微会有点长
giggle index -i "human_hm_sort/*.gz" -o human_hm_index -f -s
#这个进行的并不是特别的顺利,错误百出,以及不知道昨晚发生了什么,我的电脑自动的关机了。
(base) [xxzhang@cu08 giggle_workplace]$ giggle index -i "human_hm_sort/*.gz" -o human_hm_index -f
Segmentation fault (core dumped)
(base) [xxzhang@cu08 giggle_workplace]$ giggle index -i "human_hm_sort/*.gz" -o human_hm_index -f -s
Could not open file 'human_hm_sort/3651_sort_peaks.narrowPeak.bed.gz'
giggle: Could not open human_hm_sort/3651_sort_peaks.narrowPeak.bed.gz.
作者这个源代码是用c写的,看到源代码中有“Could not open……”的报错信息,但是根本就不知道该如何解决?我觉得作者写的太泛了,让人很没有头绪。
(吐槽)
2、比对测试
#首先使用全部的重复序列的信息进行比对。

