ADNI数据
数据的模态有
Clinical Data(临床数据)
Genetic(基因数据)
MRI
PET
BIOSPECIMEN(生物样本)
各模态数据的内容、特点
Clinical Data
内容: 招聘、人口统计、体检和认知评估数据。完整的临床数据集可以作为逗号分隔值(CSV)文件批量下载
基因数据
内容: 受试者的基因分型和测序数据,数据格式:CSV,VCF,BAM
基因分型数据:
APOE Genotyping -- CSV
TOMM40 PolyT Variant -- CSV
全基因组测序数据:
WGS (GATK Call) SNV + Indel -- VCF
WGS (CASAVA Call) SNV -- VCF
Sequenced alignment data -- BAM(不可直接下载)
存在VCF数据,不过数据量都较大,是以G为单位的
VCF数据完整的表现应为:
Record(CHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO, FORMAT, sample_indexes, samples=None)
其中:
- CHROM:染色体名称,类型为str
- POS:位点在染色体上的位置,类型为int
- ID:一般是突变的rs号,类型为str。如果是‘.’,则为None
- REF:参考基因组在该位点上的碱基,类型为str
- ALT:在该位点的测序结果。是_AltRecord类的子类实例的列表。类型为list。_AltRecord类有4个子类,代表了突变的几种类型:如snp,indel,structual variants等。所有的实例都可以进行比较(仅限于相等的比较,没有大于小于之说),部分子类没有实现str方法,也就是说不能转成字符串
- QUAL:该位点的测序质量,类型为int或float
- FILTER:过滤信息。将FILTER列按分号分隔形成的字符串列表,类型为list。如果未给出参数则为None
- INFO:该位点的一些测试指标。将‘=’前的参数作为键,后面的参数作为值,构建成的字典。类型为dict
- FORMAT:基因型信息。保存vcf的FORMAT列的原始形式,类型为str
现下载了一个较小的文件,数据量为:39.5 M(不知道当时是怎么找到的了),里面的信息以条为单位,其中前十记录表现为:
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=3, REF=T, ALT=[C])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=41, REF=C, ALT=[T])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=42, REF=T, ALT=[TC])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=55, REF=T, ALT=[C])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=56, REF=A, ALT=[AC])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=57, REF=T, ALT=[C, G])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=64, REF=C, ALT=[T])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=72, REF=T, ALT=[C, G])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=73, REF=A, ALT=[G])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=75, REF=G, ALT=[A])
Record(CHROM=gi|251831106|ref|NC_012920.1|, POS=93, REF=A, ALT=[G])
读取方式
import vcf
import os
vcf_file = "adni_mito_genomes.vcf"
vcf_reader = vcf.Reader(filename=vcf_file)
i = 0
for record in vcf_reader:
print(record)
if i == 10:
break
i += 1
MRI
内容: 原始、预处理和后处理的图像文件,FMRI和DTI
数据格式: MRI(structural, diffusion weighted imaging, perfusion, and resting state sequences)
可得到的图像数据
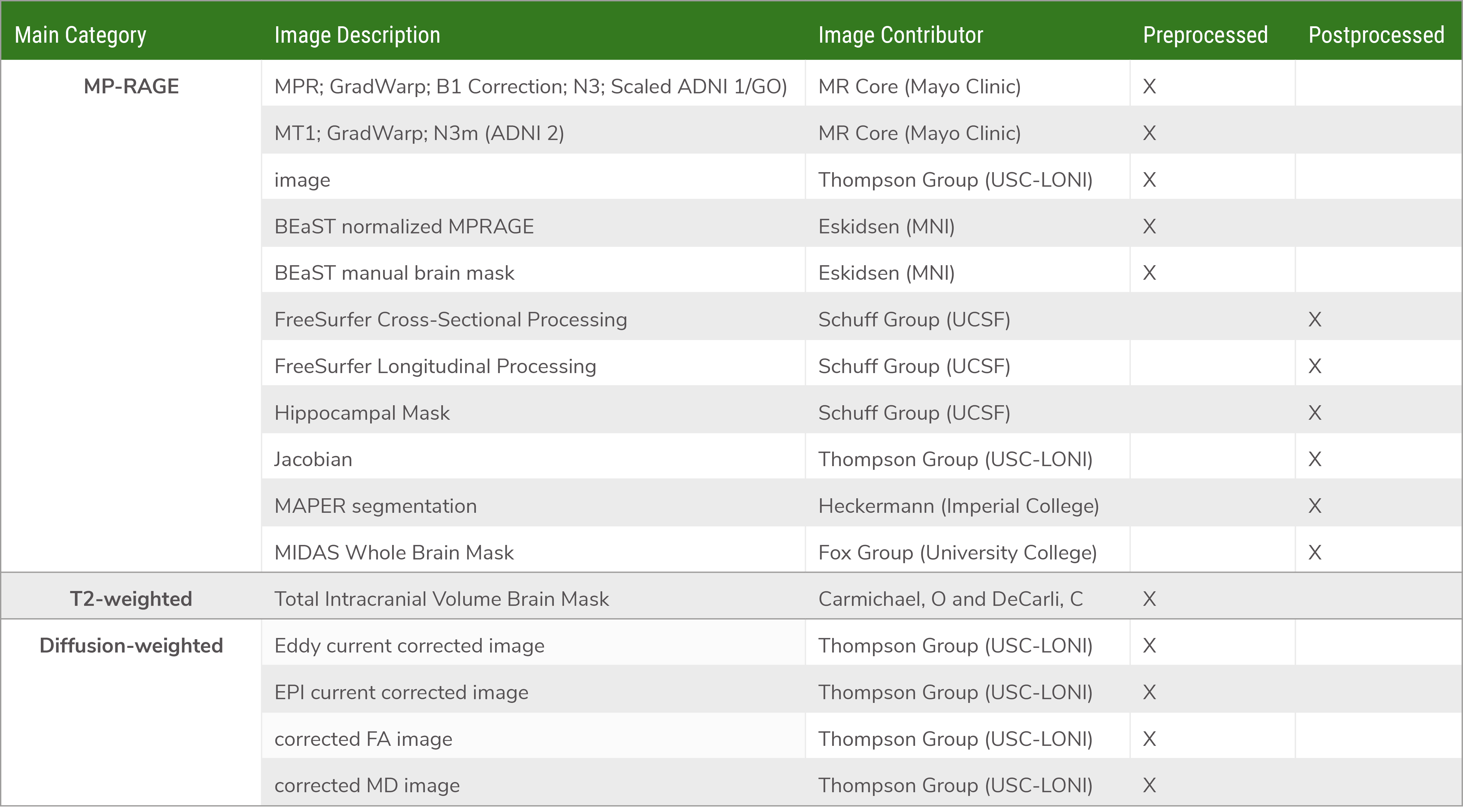
图像示例
下载数据(经过处理)示例:
名字: ADNI1_Complete_2Yr_1.5T
格式: NiFTI
大小: 22.5 M
尺寸: 192 * 192 * 160
类型: T1
制造商: SIEMENS
成像信息:
Acquisition Plane=SAGITTAL; Acquisition Type=3D; Coil=HE; Field Strength=1.5 tesla; Flip Angle=8.0 degree; Manufacturer=SIEMENS; Matrix X=192.0 pixels; Matrix Y=192.0 pixels; Matrix Z=160.0 ; Mfg Model=Symphony; Pixel Spacing X=1.25 mm; Pixel Spacing Y=1.25 mm; Pulse Sequence=IR/GR; Slice Thickness=1.2000000476837158 mm; TE=3.609999895095825 ms; TI=1000.0 ms; TR=3000.0 ms; Weighting=T1
使用Mango可直接显示图片,效果如下
名字:ADNI1_Baseling_3T
格式:NiFTI
大小:22.5 M
尺寸: 192 * 192 * 160
制造商: GE MEDICAL SYSTEMS
类型: T1
成像信息:
Acquisition Plane=SAGITTAL; Acquisition Type=3D; Coil=8HRBRAIN; Field Strength=3.0 tesla; Flip Angle=8.0 degree; Manufacturer=GE MEDICAL SYSTEMS; Matrix X=256.0 pixels; Matrix Y=256.0 pixels; Matrix Z=166.0 ; Mfg Model=SIGNA EXCITE; Pixel Spacing X=1.0156199932098389 mm; Pixel Spacing Y=1.0156199932098389 mm; Pulse Sequence=RM; Slice Thickness=1.2000000476837158 mm; TE=2.8399999141693115 ms; TI=900.0 ms; TR=6.616000175476074 ms; Weighting=T1
使用Mango可直接显示图片,效果如下

ADNI中的扫描是在两种不同的特斯拉扫描仪上进行的,即飞利浦医疗系统和西门子
飞利浦医疗系统扫描的EPI序列为144个体积,场强=3.0特斯拉,翻转角=80.0°,TE=30.0ms,TR=3000.0ms,64×65矩阵,6720.0层厚度为3.31mm的静止状态fMRI
用飞利浦医学系统扫描仪进行扩展静息状态fMRI的EPI序列为:200体积,场强=3.0tesla,翻转角=90.0°,TE=30.0ms,TR=3000.0,64×65矩阵,9600.0层厚3.31mm
对于西门子扫描仪,EPI序列是197个体积,场强=3.0特斯拉,翻转角=80.0度,TE=30.0ms,TR=2999.99,448×448矩阵,以及197个3.4mm厚度的切片
(此处显示的信息与下载的经过处理的信息TE不一致)
python代码示例
import skimage.io as io
import nibabel as nib
import numpy as np
import random
nii_file = "1.nii"
img = nib.load(nii_file)
img_arr = img.get_fdata()
img_arr = np.squeeze(img_arr)
#随机选取一张图片
img_arr1 = img_arr[:, :, random.randint(0, img_arr.shape[2])]
# 数据归一化至[0,1]
print(img_arr.shape)
img_arr1 = (img_arr1 - np.min(img_arr)) / (np.max(img_arr) - np.min(img_arr))
io.imshow(img_arr1)
io.show()
注:只能找到T1的图像,T2的未找到
PET
特点:该数据的目标是跟踪老年痴呆症的恶化和潜在的病理变化
可得到的数据
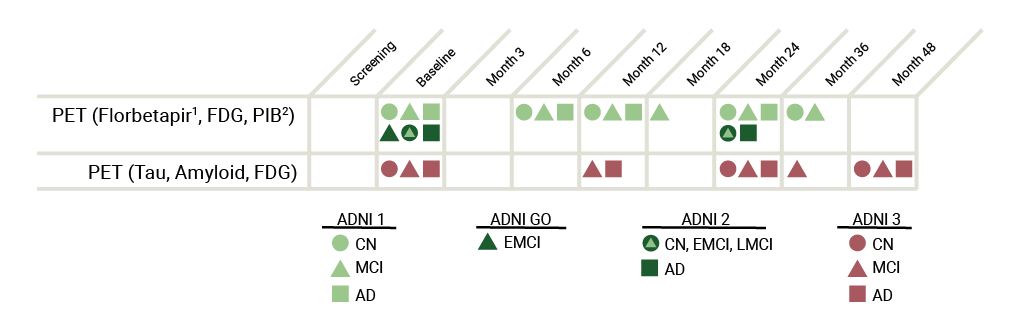
图像示例
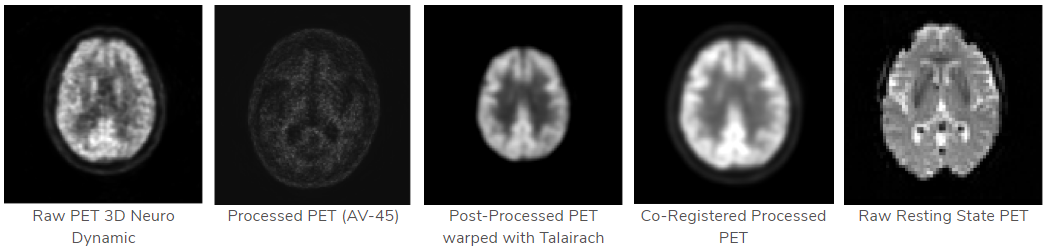
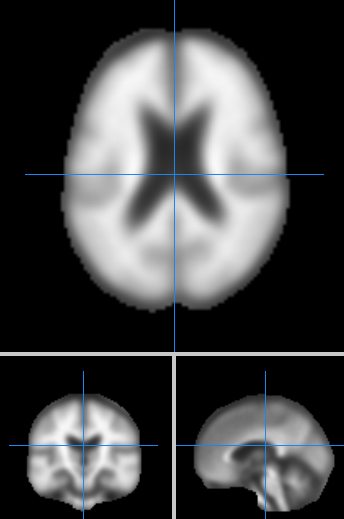
下载的数据:
单个图片大小:3.44 M
格式: NiFTI
尺寸: 91 * 109 * 91
使用Mango可直接显示图片,效果如下
BIOSPECIMEN(生物样本)
内容:血液、尿液和脑脊液(CSF)等生物标本

 浙公网安备 33010602011771号
浙公网安备 33010602011771号