


python版的MCScan绘图
最近发现了python版的MCScan,是个大宝藏。由于走了不少弯路,终于画出美图,赶紧记录下来。
github地址 https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)
-
软件安装
-
1 ## 安装lastal 2 网址:http://last.cbrc.jp 3 unzip last-1060.zip 4 cd last-1060 5 make 6 7 # 把scripts, src 添加到环境变量 8 9 ## jcvi 10 pip install jcvi 11 12 ## 若出现 from rillib.parse import urlparse 缺少parse模块,则装parse模块,然后将urllib.parse 改为urlparse; 因为urlparse模块在Python 3中重命名为urllib.parse,所以模块在Python 2.7下应该使用urlparse。 -
输入数据
-
-
-
gff文件转bed格式
-
1 ## 以spinach,和sugar为例子 2 python -m jcvi.formats.gff bed --type=mRNA --key=ID spinach_gene_v1.gff3 -o spinach.bed 3 python -m jcvi.formats.gff bed --type=mRNA --key=ID BeetSet-2.unfiltered_genes.1408.gff3.txt -o sugar.bed 4 5 ##参数 6 --type:gff文件中第三列 7 --key:type对应的第九列信息前缀 8 9 我们分析只需要用到每个基因最长的转录本就行,在sugar中是以多个转录本进行存储,因为需要获取最长转录本 10 11 ## 将sugar中bed进行去重复 12 python -m jcvi.formats.bed uniq sugar.bed
-
获取cds/pep序列
-
1 ## cds和pep序列均可以进行共线性分析 2 ## 根据上述得到的.bed文件调取对应cds和蛋白序列 3 # spinach 4 seqkit grep -f <(cut -f4 spianch.bed) spinach.cds.fa | seqkit seq -I >spinach.cds 5 seqkit grep -f <(cut -f4 spianch.bed) spinach.pep.fa | seqkit seq -I >spinach.pep 6 7 # sugar 8 seqkit grep -f <(cut -f4 sugar.uniq.bed) BeetSet-2.genes.1408.cds.fa | seqkit seq -i >sugar.cds 9 seqkit grep -f <(cut -f4 sugar.uniq.bed) BeetSet-2.genes.1408.pep.fa | seqkit seq -i >sugar.pep
-
小知识:也可以根据gff文件,基因组ref.fa文件中直接调取cds,和pep序列
-
1 ## 需要安装cufflinks 2 3 ## 提取cds 4 gffread in.gff3 -g ref.fa -x cds.fa 5 6 ## 提取pep 7 gffread in.gff3 -g ref.fa -y pep.fa
-
-
-
共线性分析
-
1 ## 新建一个文件夹,方便在报错的时候,把全部都给删了 2 mkdir cds && cd cds 3 ln -s ../sugar.cd ./ 4 ln -s ../sugar.uniq.bed ./sugar.bed 5 ln -s ../spinach.cds ./ 6 ln -s ../spinch.bed ./ 7 8 ## 运行代码 9 python -m jcvi.compara.catalog ortholog (--dbtype prot[蛋白分析]) --no_strip_names spinach sugar 10 11 结果: 12 spinach.sugar.anchors:共线性block区块(高质量) 13 spinach.sugar.last:原始的last输出 14 spinach.sugar.last.filtered:过滤后的last输出 15 spinach.sugar.pdf:点阵图 16 17 ## 如果遇到报错,多半是要安装python包,更新Latex
-
可视化
-
-
共线性图
-
1 ## 首先生成.sinple文件 2 python -m jcvi.compara.synteny screen --minspan=30 --simple spinach.sugar.anchors spinach.sugar.anchors.new 3 4 ## 编辑配置文件seqids 和layout 5 6 #设置需要展示染色体号 7 vi seqids 8 chr1,chr2,chr3,chr4,chr5,chr6 #spinach 9 Bvchr1.sca001,Bvchr2.sca001,Bvchr3.sca001 #sugar 10 11 # 设置颜色,长宽等 12 vi layout 13 # y, xstart, xend, rotation, color, label, va, bed 14 .6, .1, .8, 0, red, spinach, top, spinach.bed 15 .4, .1, ,8, 0, blue, sugar, top, sugar.bed 16 # edges 17 e, 0, 1, spinach.sugar.anchors.simple 18 19 注意, #edges下的每一行开头都不能有空格 20 21 ## 运行代码 22 python -m jcvi.graphics.karyotype seqids layout
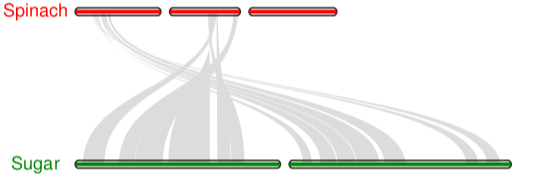
-
-
-
若要突出显示某一共线性则可以在对应的位置添加g*
-
1 vi spinach.sugar.anchors.simple 2 g*Spo03717 Spo03716 Bv3_048620_odzi.t1 Bv3_049090_cxmm.t1 46 + 3 Spo17356 Spo17350 Bv1_001140_tedw.t1 Bv1_001540_xzdn.t1 41 - 4 Spo13685 Spo13730 Bv5_123480_yfcy.t1 Bv5_123900_rucq.t1 46 - 5 Spo26250 Spo26280 Bv5_117270_qhwj.t1 Bv5_117680_iykf.t1 36 + 6 Spo19005 Spo06982 Bv7_173830_frmo.t1 Bv7_174150_pckw.t1 37 + 7 Spo19374 Spo20559 Bv4_081440_riqq.t1 Bv4_081750_yeuy.t1 32 - 8 9 #运行 10 python -m jcvi.graphics.karyotype seqids layout
-
-
-
若要显示3个物种的共线性,则应两两比对,得到两个*.simple文件,并对其进行配置(来自https://www.jianshu.com/p/39448b970287)
-
1 $ vi layout 2 # y, xstart, xend, rotation, color, label, va, bed 3 .7, .1, .8, 15, , Grape, top, grape.bed 4 .5, .1, .8, 0, , Peach, top, peach.bed 5 .3, .1, .8, -15, , Cacao, bottom, cacao.bed 6 # edges 7 e, 0, 1, grape.peach.anchors.simple 8 e, 1, 2, peach.cacao.anchors.simple 9 10 $ vi seqids 11 chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19 12 scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8 13 scaffold_1,scaffold_2,scaffold_3,scaffold_4,scaffold_5,scaffold_6,scaffold_7,scaffold_8,scaffold_9,scaffold_10r 14 15 $ python -m jcvi.graphics.karyotype seqids layout
-
-

-
-
-
3个物种三角形排序配置文件(来自https://www.jianshu.com/p/f7971dbf5f85)
-
1 # y, xstart, xend, rotation, color, label, va, bed 2 .5, 0.025, 0.625, 60, , Grape, top, grape.bed 3 .2, 0.2, .8, 0, , Peach, top, peach.bed 4 .5, 0.375, 0.975, -60, , Cacao, top, cacao.bed 5 # edges 6 e, 0, 1, grape.peach.anchors.simple 7 e, 1, 2, peach.cacao.anchors.simple 8 9 #运行 10 python -m jcvi.graphics.karyotype seqids layout
-
-

除此之外,Tbtools也可以完成类似图片,对于新手来说更加容易上手,具体可查看以下内容
参考
1、其实MCScan画图也可以很好看
2、「JCVI教程」如何绘制CNS级别的共线性图(上)
3、「JCVI教程」如何绘制CNS级别的共线性图(中)
关注下方公众号可获得更多精彩


