
RNA-seq 生物学重复相关性验证
根据拿到的表达矩阵设为exprSet
1、用scale 进行标准化
数据中心化:数据集中的各个数字减去数据集的均值
数据标准化:中心化之后的数据在除以数据集的标准差。
在R中利用scale方法来对数据进行中心化和标准化
1 scale(data, center=T, scale=F) 2 3 其中,center为T,表示数据中心化 4 5 scale为T,表示数据标准化 6 7 对一个data frame的每一列进行计算
并不是表达矩阵里面的所有基因都可以进行相关性分析,首先去除reads count >1 小于5个的基因(测试样品共有6个)
1 ##过滤reads count >1 小于5 2 exprSet <- exprSet[apply(exprSet,1, function(x) sum(x>1) >5,] 3 ##reads count 差距较大,用log以及scale 缩小差距 4 M <- scale(cor(llog2(exprSet+1))) 5 ##热图 6 pheatmap(M)
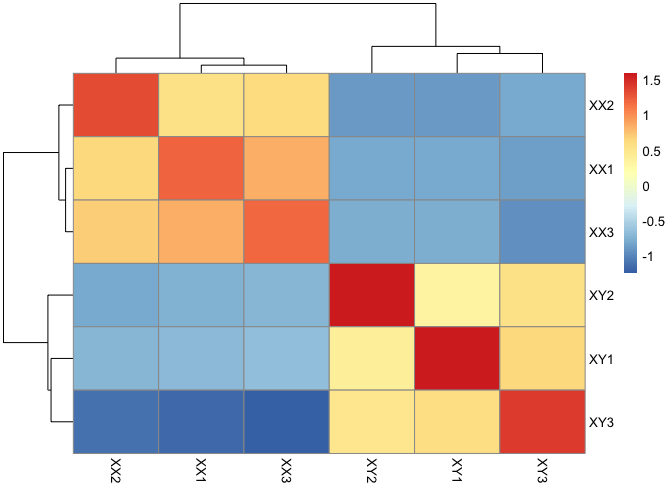
2、另一种标准化
1 exprSet <- exprSet[apply(exprSet,1, function(x) sum(x>1) >5,] 2 3 ##去除文库大小差异 4 exprSet <- log(edgeR::cpm(exprSet)+1) 5 6 ##取mad(绝对中位差)(类似sd)的前50% 7 exprSet <- exprSet[names(sort(apply(exprSet,1,mad),decreasing = T)[1:500]),] ##取前500 8 M <-cor(log2(exprSet+1)) 9 10 ##添加group_list 11 tem = data.frame(g=group_list) 12 rownames(tem) <- colnames(M) 13 pheatmap::pheatmap(M, annotation_col = tem,filename = 'cor.png')
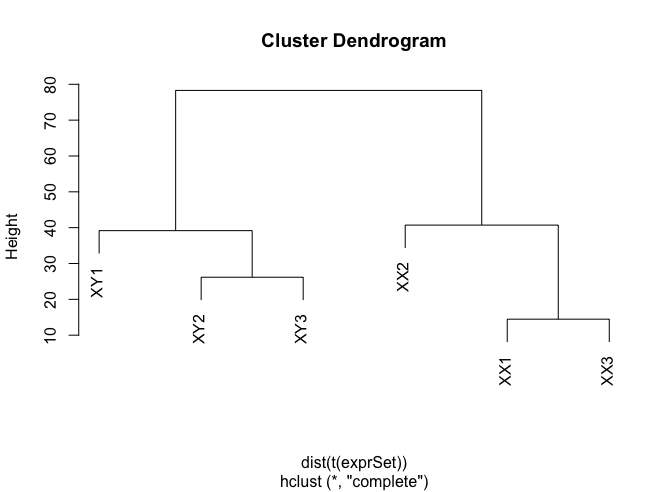
3、hclust 聚类分析
欧式距离(Euclidean Distance)
欧式距离是最易于理解的一种距离计算方法,源自欧式空间中两点间的距离公式。
用R语言计算距离主要是dist函数。若X是一个M×N的矩阵,则dist(X)将X矩阵M行的每一行作为一个N维向量,然后计算这M个向量两两间的距离。
表达矩阵是每一行为基因,列为样品名称,所以要进行转至才能计算每个样品之间基因表达量的距离
为了得到更好的聚类分析,也可以将表达矩阵标准化,譬如,log,或者 scale等
1 hc <- hclust(dist(t(exprSet)))
2 plot(hc)
参考:生信技能树
