


序列比对和构建进化树(clustalw和phylip)
安装clustalw很简单,不提了。
找了几个蛋白序列进行比对,命名为dm.fasta

1、输入 ./clustalw2 进入交互模式

2、选择1 并输入文件名字

3、输入2, 进行多序列比对

4、如果要修改输入格式,则点9

5、若要输出格式为phylip,则点4,并关闭1
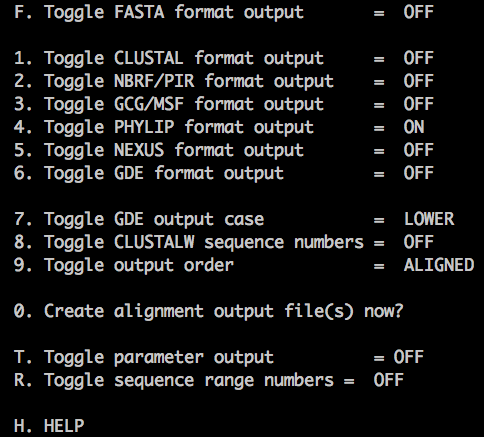
6、按下回车,后退
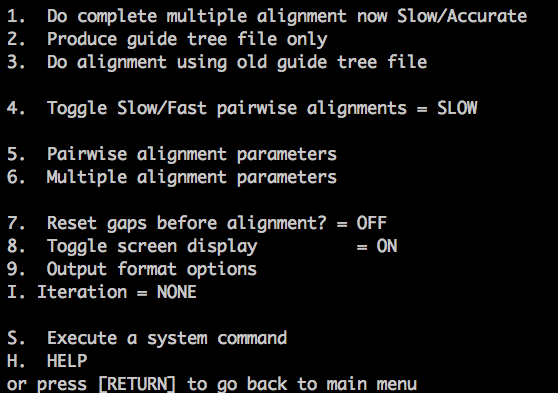
7、选择1进行比对, 因为phylip输入文件为名infile, 所以这里直接改名字infile,并退出软件即可
安装phylip
减压后,进入src 并输入 make -f Makefile.unx install
然后进入exe, 并将刚才比对结果infile 移到exe中。
进行构建树:
1、最大似然
直接输入./proml , 输入y进行确定参数,得到两个文件,outtree 和outfile,若想图形化,则将outtree 改为outtree.tre 可在mega上查看
2、临接法
先输入./protdist. 计算各个序列中两两序列的距离,得到距离矩阵。将该结果文件改名为infile,并进行临接法构进化树,方法为;输入./neighbor.用同样的方法可以在mega上查看图像
关注下方公众号可获得更多精彩

