NGS的duplicate的问题
duplicate的三个问题:
一.什么是duplicate?
二.duplicate来源?
三.既然PCR将1个reads复制得到成百上千copies,那为什么二代数据duplicate rate 一般才10+%?
什么是 duplicate?
摘自罗俊峰博士,阅尔基因研发总监陈云地博士,阅尔基因CTO http://www.biotrainee.com/thread-1382-1-1.html
一、什么是Duplicated Reads
1
谈到NGS数据的duplicated reads(暂且翻译为“重复数据”),我们通常会直观地认为:duplicated reads是在NGS文库构建过程中,由于PCR过度扩增导致同一个模板DNA片段被反复测序多次,得到一模一样的reads。
2
但是这经不起推敲。仔细一想,就很困惑。
PCR不就是用来产生重复数据的吗?否则不叫PCR了。除了PCR-free的文库构建方法以外,大部分NGS文库构建方法都有PCR步骤,难道说这些NGS数据都有问题?
这是不可能的。或许:
PCR可以产生重复序列,但是不能额外多产生一条或多条。设一个基因组有A、B两个片段,PCR后,如果得到1000A+1000B,是正确的;如果得到1000A+1000A+1000B,多出来的1000A就是重复数据?问题是,PCR怎么会凭空多出来1000条片段A的测序reads呢?这要PCR出了什么样的问题,才能产生出这样的结果?
PCR是不会这样的。或许:
A+B经过PCR后得到1500A+1000B,多出来的500条A是重复数据?这不就是大家常说的PCR bias吗?
到底什么是“过度扩增”呢?
3
严格的定义是这样的:
duplicated reads是PCR对同一个分子进行多次镜像复制的后果。
判断是否为镜像分子的标准是:reads的起始和终止位置一样,起点和终点之间的碱基序列一样(不妨简称为“三一样”)。只要起点、终点、或者起点与终点之间的序列三者之中有一个不同,就是不同的分子,称为unique reads。
镜像复制出来的分子个数与总分子数的比例就是duplication rate,duplication rate = 1 - unique reads / total reads。
4
PCR本来就是用来镜像复制DNA片段的。对于最理想的NGS数据分析,难道要尽可能把所有通过PCR获得的子链的测序数据全部去除,要把PCR的效果完全消除,要还原到没有PCR的状态?
是的。
设一个基因组有A、B两个片段,PCR后得到无论多少条reads,比如n・A+m・B条,在数据分析的时候,都只保留1条A和1条B(unique reads)用于组装,而去掉(n-1)条A和(m-1)条B。共有(n-1)条A和(m-1)条B被当成duplicatedreads看待,尽管它们是正常PCR的正常产物。
所以,
目前的算法其实是一个简化的处理方案,把所有重复的reads都去掉了,留下完全不重复的reads。算法没有能力区分“假重复”(人为造成的重复序列方面的bias)和“真重复”(天然存在的重复序列)。
所以,
对于NGS 数据而言,Duplicateddata是一个生物信息学概念,不是分子生物学概念;是人为规定的,不是文库构建、高通量测序等生化反应自然生成的。
以下摘自 wangpeng905 链接:https://www.jianshu.com/p/1e6189f641db
为什么会有 duplicate?
要弄清楚这个问题,需要从 NGS 数据产出流程说起:
- 基因组核酸提取
- 基因组 DNA 随机打断,最常用的是超声打断。
- 被打断的 DNA 片段经历末端修复,3' 加A,两端加接头,选择特定大小片段文库进行 PCR 扩增(通过 PCR 扩增选择性提高加上了接头的文库分子数量)。
- 文库上机与 flowcell 上引物结合,经历桥式 PCR 扩增形成 cluster 。
- 进行 SBS 测序,光学信号捕获,生成序列。
我们首先假设基因组核酸提取是完整的基因组,打断是完全随机的(通常是这样的)。
在第 3 步,PCR 扩增时同一个文库分子会产生多个相同的拷贝,这是 duplicate 的主要来源(PCR duplicate)。
第 4 步,文库中 DNA 片段与 flowcell 上引物结合,来源于同一个 DNA 片段的多个拷贝都结合到 flowcell 上,这样会导致生成多个相同的 cluster,测序时也就有多个相同的序列被测出来,这些相同的序列就是 duplicate。
同在第 4 步,生成 cluster 时候一个 cluster 中的 DNA 链可能搭到旁边另外一个 cluster 生成位点上,又长成一个相同的 cluster ,这也是 duplicate 的一个来源(Hiseq4000之后的 flowcell 会有的 cluster duplicate)。
第 5 步,一个 cluster 测序时的捕获的荧光亮点由于形状奇特,可能被软件当成两个荧光点来处理,这也产生了两条完全相同的 reads。这个过程中可能产生完全相同的 reads。(光学 duplicate)
由此我们知道,PCR duplicate 特点是随机分布于 flowcell 表面,光学 duplicate 特点是它们都来自 flowcell 上位置相邻的 cluster 。cluster 的位置被记录在 Fastq 文件 @seq-id 这一行中。
下图的右下角还有一种 duplicate 来源,sister? 这种一个文库分子的两条互补链同时都与 flowcell 上的引物结合分别形成了各自的 cluster,最后产生的两对 reads 完全反向互补,map 到参考基因组也分别在正负链上的相同位置,有的分析中也算 duplicate,虽然我遇到的这种正负链测序结果通常是不算 duplicate 的。
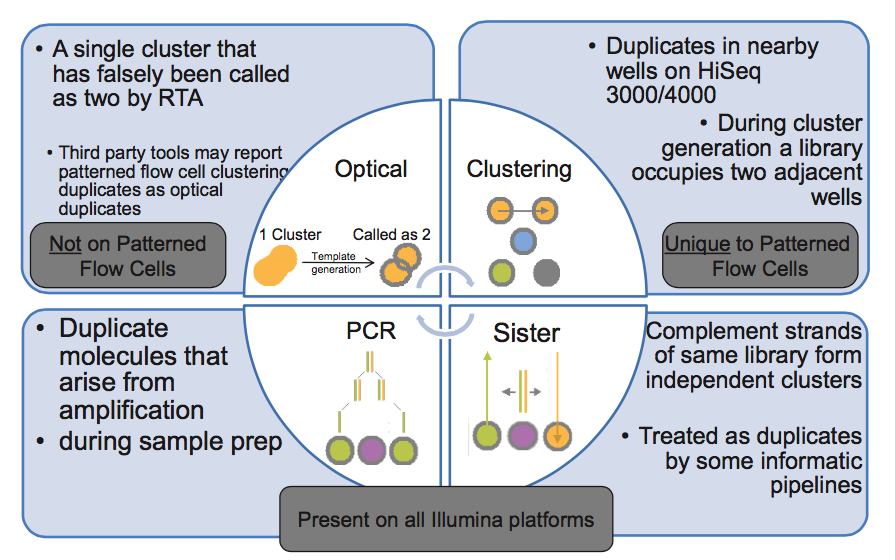
另外,据说 NextSeq 平台上出现过由于荧光信号捕获相机移动位置不够,导致 tile 边缘被重复拍摄,每次采样区域的边缘由于重复采样而出现的 duplicate,下图中蓝色点代表 duplicate ,在 tile 两侧明显富集。Illumina 公司回应说这没毛病,符合预期……
PCR 将模板扩增了数千倍,但数据中 duplication 率只有 15%
我曾经有这样的疑惑,为什么文库构建过程中的 PCR 将每个文库分子都扩增了上千倍,以 PCR 10个循环为例 2^10= 1024 ,但是实际测序数据中 duplication 率并不高(低于20%)。后来我看到一篇文章从统计概率的角度详细探讨了一下 duplication 率的影响因素,顺便一提,这个博主的故事也很令人佩服。
PCR 的过程中不同长度的文库分子被扩增的效率不同(GC 太高或 AT 含量太高都会影响扩增效率),PCR 更倾向于扩增短片段的文库分子,这里先不考虑文库片段扩增效率的差异,把问题简化一下,假设所有文库分子扩增效率都相同。PCR duplicate 的主要来源是同一个文库分子的不同拷贝都在 flowcell 上生成了可以被测序的 cluster ,导致同一个分子的序列被测序仪读取多次。那么为何在每个分子都有上千个拷贝的情况下,实际却很少出现同一分子的多个拷贝被测序的情况呢?主要原因就是文库中 unique 分子的数量比被 flowcell 上引物捕获的分子数量多很多,直白点说就是 flowcell 上用于捕获文库分子的引物数量太少了,两者不在同一个数量级,导致很少出现同一个文库分子的多个拷贝被 flowcell 上引物捕获生成 cluster。
假设文库中所有分子与引物的结合都是随机的,简化一下就相当于,一个箱子中有 n 种颜色的球(文库中的 n 种 unique 分子),每种颜色有 1000 个(PCR 扩增的,随 cycle 数变化),从这个箱子中随机拿出来 k 个球(最终测序得到 k 条 reads),其中出现相同颜色的球就是 duplicate,那么 duplication 率就可以根据有多少种颜色的球被取出 0,1,2,3…… 次的概率计算,可以近似用泊松分布模型来描述。
以人全基因组重测序 30X 为例,PE150 需要约 3x10^8条 reads ,文库中 unique 分子数其实可以通过上机文库的浓度和体积(外加 PCR 循环数)计算出来,这里用近似值 3.5x10^10 个 unique 分子。每个 unique 分子期望被测序的次数是 3x108/3.5x1010 = 0.0085 ,每个 unique 分子被测 0,1,2,3… 次的概率如下图:
> x <- seq(0,10,1)
> xnames <- as.character(x)
> xlab <- "一个文库分子的所有拷贝被测序的次数"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.0085),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)
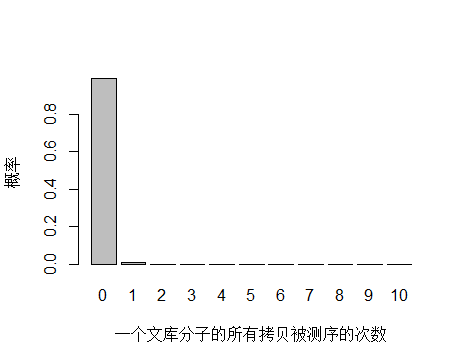
由于 unique 分子数量太多,被测 0 次的概率远高于 1 和 2 次,我们去除 0 次的看一下:
> x <- seq(1,10,1)
> xnames <- as.character(x)
> xlab <- "一个文库分子的所有拷贝被测序的次数"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.0085),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)
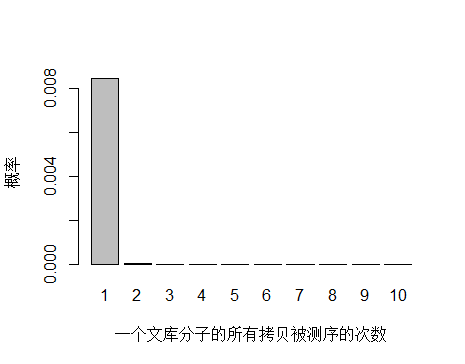
unique 分子被测序 1 次的概率远大于 2次及以上,即便一个 unique 分子被测序 2 次,我们去除 duplicate 时候还会保留其中一条 reads。
如果降低文库中 unique 分子数量到 4.5x10^9 个,PCR 循环数增加以便浓度达到跟上面模拟的情况相同,测序 reads 数还是 3x10^8 条,每个 unique 分子预期被测序的次数是 3x108/4.5x109 = 0.067 。
> x <- seq(1,10,1)
> xnames <- as.character(x)
> xlab <- "一个文库分子的所有拷贝被测序的次数"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.067),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)
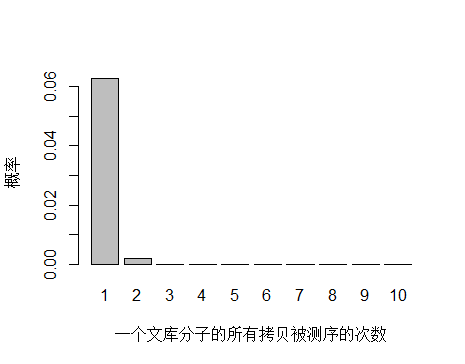
unique 分子数量减少,被测序 2次的概率增大,duplication 率显然也会增高。
到这里已经可以很明白的看出 duplication 率主要与文库中 unique 分子数量有关,所以建库过程中最大化 unique 分子数是降低 duplication 率的关键。文库中 unique 分子数越多,说明建库起始量越高,需要 PCR 的循环数越少,而文库中 unique 分子数越少,说明建库起始量越低,需要 PCR 的循环数越多,因此提高建库起始量是关键。
作者:wangpeng905
链接:https://www.jianshu.com/p/1e6189f641db

 浙公网安备 33010602011771号
浙公网安备 33010602011771号