搭建本地NCBI病毒库用于Blast
搭建本地NCBI病毒库用于Blast
目的:为了通过Blast剔除我数据集中所有与Human任意片段相似度超过97%的序列
日期:2022/11/17
1. Nt库下载
创建conda环境
conda create -n aspera
conda activate aspera
conda install -y -c hcc aspera-cli
conda install -y -c bioconda sra-tools


下载Nt库
# 在/media/yang/data/nt目录下下载nt.gz
ascp -v -k 1 -T -l 200m -i ~/miniconda2/envs/rna/etc/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/blast/db/FASTA/nt.gz ./
# 然后在/media/yang/data/nt目录下下载nr.gz
ascp -v -k 1 -T -l 200m -i ~/miniconda2/envs/rna/etc/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/blast/db/FASTA/nr.gz ./
2. 软件准备
2.1、数据:
-
accession2taxid:(核酸就下载核酸,蛋白就下载蛋白)https://ftp.ncbi.nih.gov/pub/taxonomy/accession2taxid/https://ftp.ncbi.nih.gov/pub/taxonomy/accession2taxid/nucl_gb.accession2taxid.gz
-
taxdump: https://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
2.2、软件:
sudo cp taxonkit /usr/local/bin/
2.3、解压
gzip -d nr.gz
mv nr nr.fa
gzip -d prot.accession2taxid.gz
gzip -d taxdump.tar.gz
把taxdump解压的文件移动到taxonkit对应位置,否则会报错
sudo cp *.dmp /home/yzu/.taxonkit
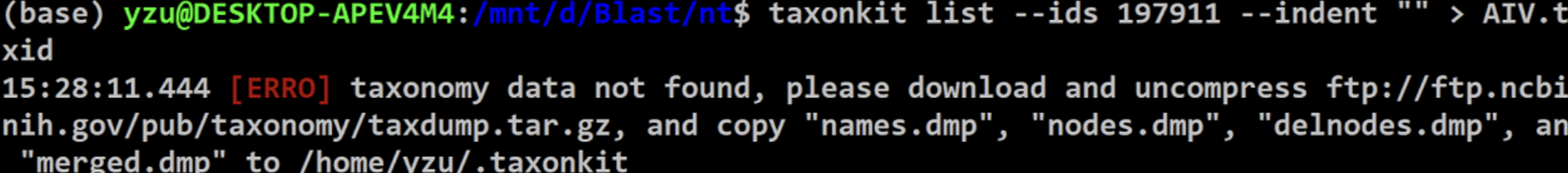
3. 抽提所有的流感序列构建子库
3.1 nt本地库构建(可有可无)
makeblastdb -in nt.fa -dbtype nucl -title nr -parse_seqids -hash_index -out nr -logfile nr.log
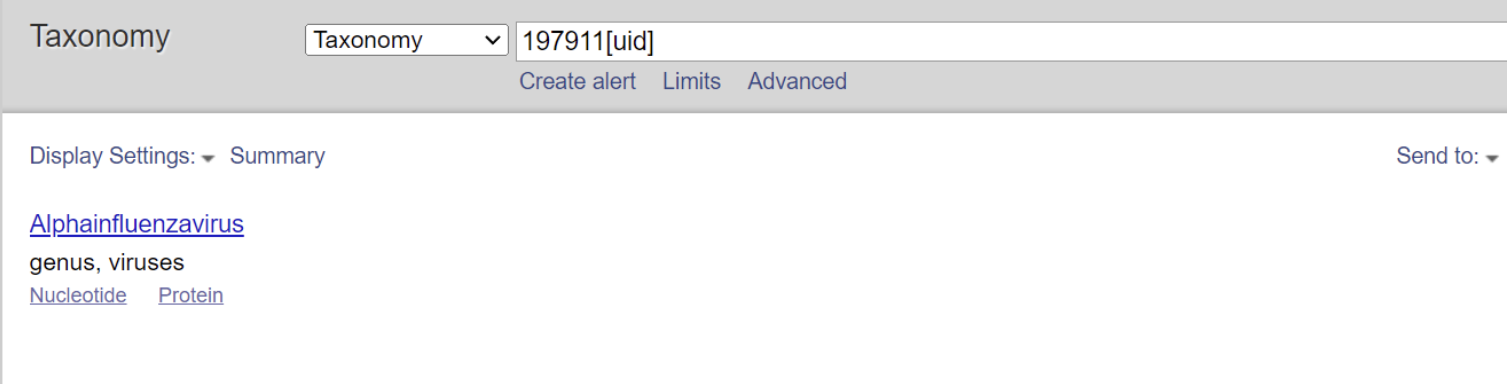
A型流感病毒对应的taxonomy id是197911
3.2 抽取所有的AIV序列
taxonkit list --ids 197911 --indent "" > AIV.taxid
cat prot.accession2taxid |csvtk -t grep -f taxid -P AIV.taxid |csvtk -t cut -f accession.version > Viruses.acc
seqtk subseq nt.fa Viruses.acc > IAV.fa
3.3 建索引
makeblastdb -in IAV.fa -dbtype nucl -title NCBIIAV -parse_seqids -hash_index -out NCBIIAV
4. Blast
sudo blastn -task blastn -db NCBIIAV -query /mnt/c/Users/yzu-v/Desktop/all_avian.fas -outfmt 7 -out query.txt
获得更为纯净的blast结果
sed '/^#/d' query.txt > query_result.txt
只筛选相似度大于97%的结果
awk '$3 >= 97 {print}' query_result.txt > query_to_rm.txt
5. 登录号转name
本文的目的是找到所有和人源毒株有某一个片段相似度大于97%的毒株,所以要把登录号转为name,这样更容易看结果。
我们需要用excel制作对应的accession→name表格,但是前文提到了,我们建索引的nt库的IAVs子集也非常大,有50多W条序列,这样我们就需要用python来提取fasta序列的id和对应的name,否则是会卡死的。
from Bio import SeqIO
import pandas as pd
seqid = []
seqname = []
for seq_record in SeqIO.parse("IAV.fa","fasta"):
seqid.append(seq_record.id)
seqname.append(seq_record.description)
dict_1 = {
"id": seqid,
"name": seqname
}
data = pd.DataFrame(dict_1)
data.head()
data.to_csv("acc_to_name.csv")
接下来就去excel里面分列做一个自己感兴趣的信息表就行了,最后我就提取一下包含human字符的所有结果行就行了
grep 'human' merge.csv > to_rm.csv

 浙公网安备 33010602011771号
浙公网安备 33010602011771号