

生物信息学
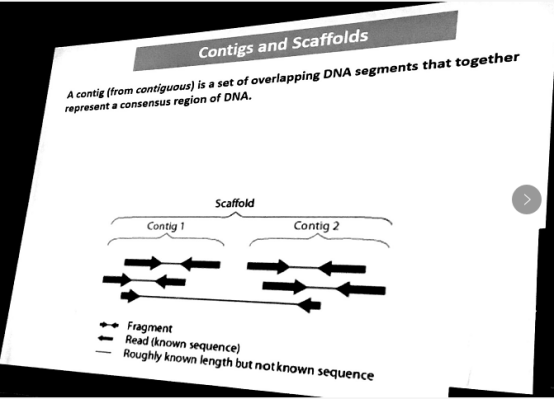
Contig是reads拼成的连续的DNA片段,连续表达一个gene。通过双端测序的contig可确定contig之间的关系得到scaffold,Scaffold是reads拼成的有gap的DNA片段。理想情况下,一条染色体用同一个scaffold的表达。整个genome存在很多零碎片段,可舍弃。因为duplication产生很多overlap。
N50,L50和NG50是评价genome assembly的quality的标准,评价长度时使用N50,N50是一个contig的长度。不选用genome size的50%是因为1.这是估计的size值不一定准;2.sequence 仅覆盖80%。评价数量使用L50,L50数量越小越好。NG50表达测到genome 覆盖度。

取材方法很重要,得到目标数据。
Assembly算法有可能带来更多误差,通常二代测序和三代测序相结合。


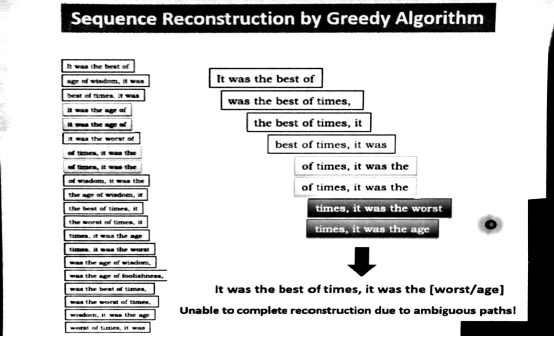
贪心算法原理是每一步都在找最优解,最后得到最好的结果,优点是快,缺点是不是全局最优解,出现重复序列便走不下去。

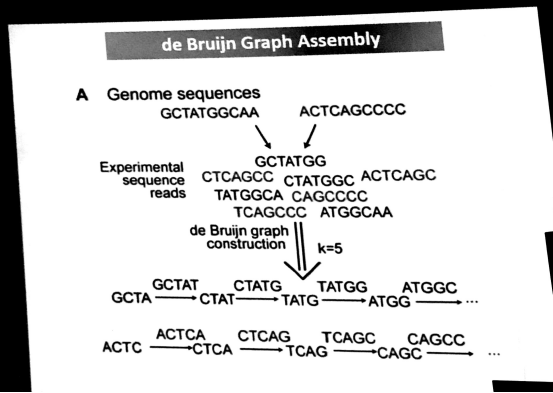
de bruiji graph来自桥问题:
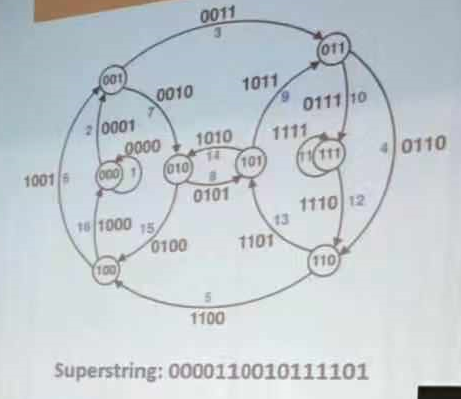
比如:
k-mer当k=4时
000添0,成为0000,0000取000,若下一步添0 成为0000
000添1,成为0001,0001取001,若下一步添0 成为0010,0010取010
000添1,成为0001,0001取001,若下一步添1 成为0011,0011取011
在序列拼接时:
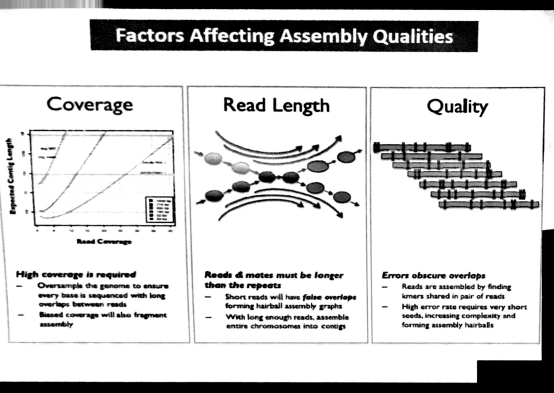
Qualities取决于二代测序的质量;coverage&read length取决于建库方法和sequence方法。
生物信息学处理关键是考虑研究的物种的特性,eg:某物种的duplication多,或者生物学问题的侧重点,eg:重测序。
Genome网站:UCSC
例子:
植物类,希望通过“拟南芥vs抗逆抗旱植物”,清楚看到抗逆抗旱植物的相关特性,或者说希望看到的特性,做之前要估算genome size,可以看的现象是扩增基因,于是分析扩增基因的具体情况,比如对于常规部分的不同功能、通路及转录因子等,对于miRNA的探讨,分析出现的原因。
以前的研究多重于分析生物学特征,比如某物种所有基因的罗列,现在基因组数量变大之后多研究.进化特征,比如某一个基因,eg:所有种类黄瓜的苦味,这种主观感觉定量分析的研究。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号