

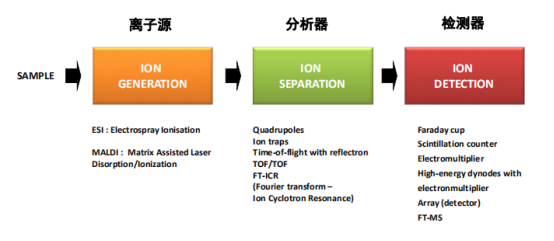
质谱仪:
质谱分析法是先将大分子电离为带电粒子,按质核比分离,由质谱仪识别电信号得到质谱图。
Top-down直接得到结果是蛋白。
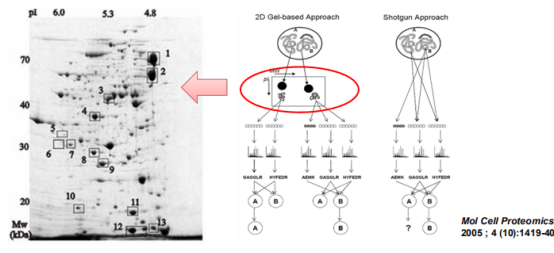
Bottom down使用shutgun方法得到结果是肽段。

由蛋白质混合物打断为肽段混合物,按特定时间分离为LC,

初次得到的谱图为一级谱,一级谱是串联质谱,其中一个峰一个肽段。选出一个峰作为二级谱的原料,选出母离子(母离子就是整个肽段 PEPMASS)并打断送入质谱仪,二级谱是一张图中的一个肽段。除此之外还可以做更多等级的图谱,tandem mass spectrometry),二级谱图用于核心定性。利用蛋白酶的对在不同理化条件下产生不同切点的性质进行酶切,一般用一种酶切,有时候为了比对用两种。其中,子离子与肽段的关系:子离子构成肽段,即子离子的构造是“羧基----离子---氨基”式组合。当AA一个个掉掉下来之后,测量得到b+y成对数据,即peptide fragment。
质谱分析方法有从头分析法和数据库搜索匹配法,常用数据库法,即实验谱图在已知肽段理论图谱数据库中找匹配。
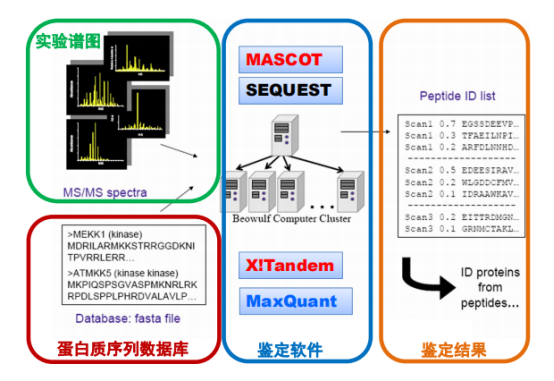
数据处理流程:

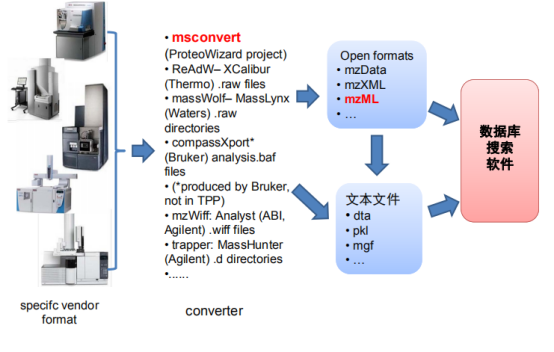
因为不同质谱仪产出不同格式,所以要对数据进行格式转换。Msconvert用于质谱转换用于预处理,可以降噪音,母离子矫正和输出规范化数据。
可以选择以下搜索引擎,Mascot的鉴定效果好;SEQUEST是传统的工具,不易学习和升级但是免费开源;X!Tandem可随需改动;pFind由小团队制作,但是灵敏度好;MaxQuant可同时产出定性和定量数据,使用方便,但覆盖率低。不同软件结果有不同,最好比对后选择一种。
可以选择数据库,Uniprot中的UniprotKB&Swiss-prot高质量,低冗余而且是人工的专家确认;neXtprot存储有关人类蛋白质数据,并有与实验证据相联系;IPI的稳定性比较差;NCBI中的RefSeq&nr是常见数据库直接加和,所以该数据库冗余量大,噪音非常大。
搜索引擎参数设定

蛋白质的鉴定和质检:
基于数据库法:
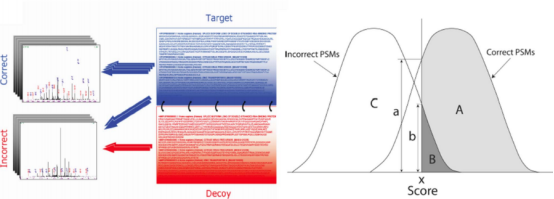
基于图谱数据库法的质检主要关注假阳性FDR false discover rate,因为从谱图到肽段到蛋白误差逐渐放大。如图,在某一图谱总错误导致一个蛋白错误。所以在谱图阶段就应该质检。原始数据中连续y连续b是好的。

先将蛋白质序列切成肽段序列,之后按照肽段分子排序,确定一个长度标准作为可鉴定出来的肽段,然后保留可鉴定长度的肽段。根据这些肽段找在数据库中候选图谱,组成理论图谱基于打分函数(Xcorr & △cn第一差距与第二差距)的得分判断图谱的可信度。
可基于正伪序列库方法使用Percolator做质量控制,因为理论图谱是正确序列和错误序列的集合,而伪库就是错误序列的集合,其补集是正取序列。如此做减法,得到图谱的线性模型或曲线模型,变成分类问题,以此找到正取的那部分。
此时存在共享肽段存在蛋白推导的困难,可以使用简约组装蛋白法。简约组装蛋白是选择最多信息承载的最少蛋白组合而成的蛋白组。常用MAYU法。

基于从头预测法:
从头测序预测方法是每个AA都算一遍,此方法可产生新蛋白,有PEAKS,NovoHMM,常用Nover方法,因其速度快,DeepNovo是深度学习方法。

