

生物信息学
Sanger采用链终止法进行测序
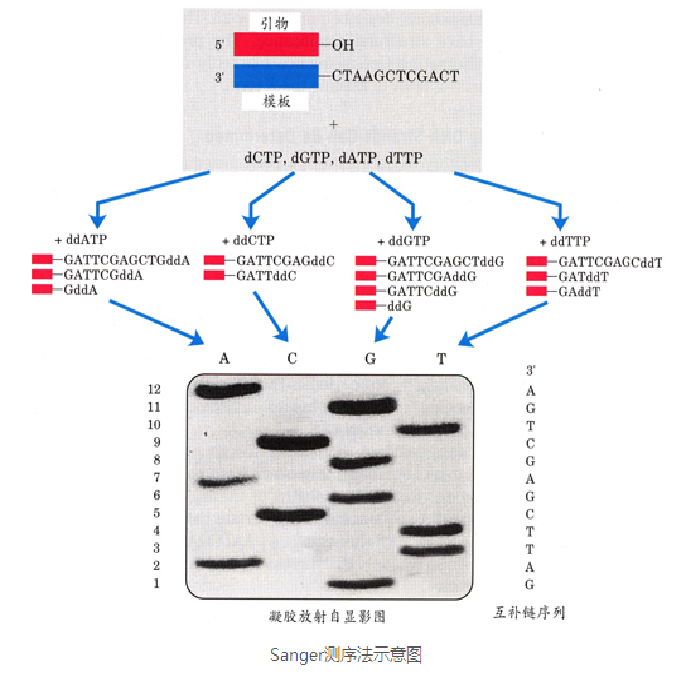
带有荧光基团的ddXTP+其他四种普通的脱氧核苷酸放入同一个培养皿中,例如带有荧光基团的ddATP+普通的脱氧核苷酸A、T、C、G放入同一个培养皿,以此类推,存在4种不同类型碱基的识别机制,同时,该ddXTP一旦结合在互补链上则会迫使复制停止。
高通量测序是二代测序,先建库后测序:
建库方法:
单末端测序:将DNA双链打碎并接上接头序列,通过改变条件使双链变单链,将待测的单链固定在flowcell上,再加入游离的脱氧核苷酸,采用边合成边测序方法比配并测得互补链,最后冲走互补链。
双末端测序:将DNA双链打碎并接上接头序列,通过改变条件使双链变单链,将待测的单链固定在flowcell上,采用桥式扩增(即将模板链和互补链单链都成簇,用酶切,就只剩下模板链和两端引物),通过桥式扩增加强信号。
它的碱基识别是在链终止法的基础上,采用边合成边测序方法:
带有荧光基团的ddXTP+其他三种普通的脱氧核苷酸放入同一个培养皿中,例如带有荧光基团的ddATP+普通的脱氧核苷酸T、C、G放入同一个培养皿,以此类推,存在4种不同类型碱基的识别机制,同时,该ddXTP一旦结合在互补链上则会迫使复制停止。 模板链通过碱基互补配对原则得到互补链,该互补链停止于需识别碱基上,通过测试最后一位碱基所携带的荧光基团,从而确定模板链对应的碱基种类。在确认了该位置的碱基种类之后,会加羟基去去除荧光基团,并使得链又不断复制下去,通过识别后的照片来得到最终的序列组成。
高通量测序的特点是数据量大。
Fasta:
>序列名字
序列本身
Fastq:
@序列信息
序列本身
@序列信息
序列质量:由碱基错误率算得,每个碱基对应一个质量,将具体数值用ASCII表示出来
质量指标:Q20、Q30、Q40
基因芯片和高通量测序的比较:
基因芯片原理:
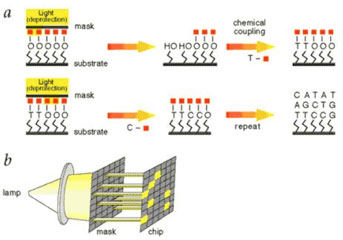
通过探针捕获游离的带有荧光基团的脱氧核苷酸,然后使其荧光标记发出荧光,辨别荧光强度确定碱基种类并加入羟基使其不再发光,再以此类推确定另外一种荧光种类。
二代测序:双末端测序;边合成边测序
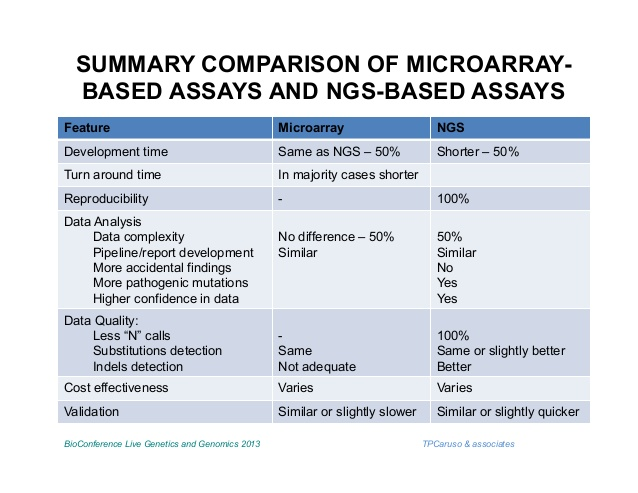
区别:前期样本制备
基因表达:相当
数据分析流程:
图片数据(识别荧光基团测序)----序列信息(fastq)------质控-----assembly----analysis----annotation
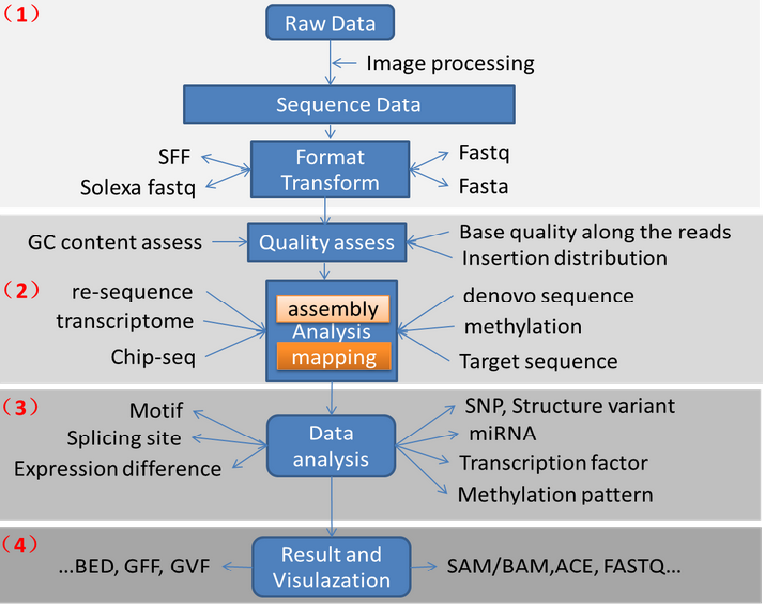
Raw data------质控fastaQC------去adoptor/linker(引物)------使用barcode排序------FASTX-Toolkit质控(某特殊位置之后的;低于平均值的)
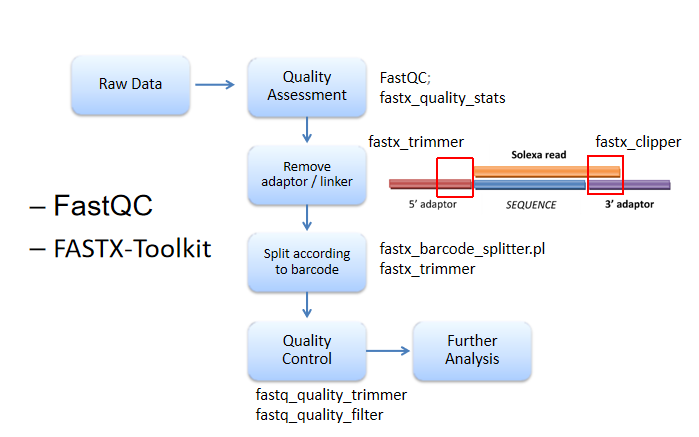
fastaQC:FASTQC checks whether a set of sequence reads in a .fastq fifile exhibit any unusual qualities.

FASTX-Toolkit:In this section of the tutorial we will be using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) to fifilter and trim sequences based on quality.
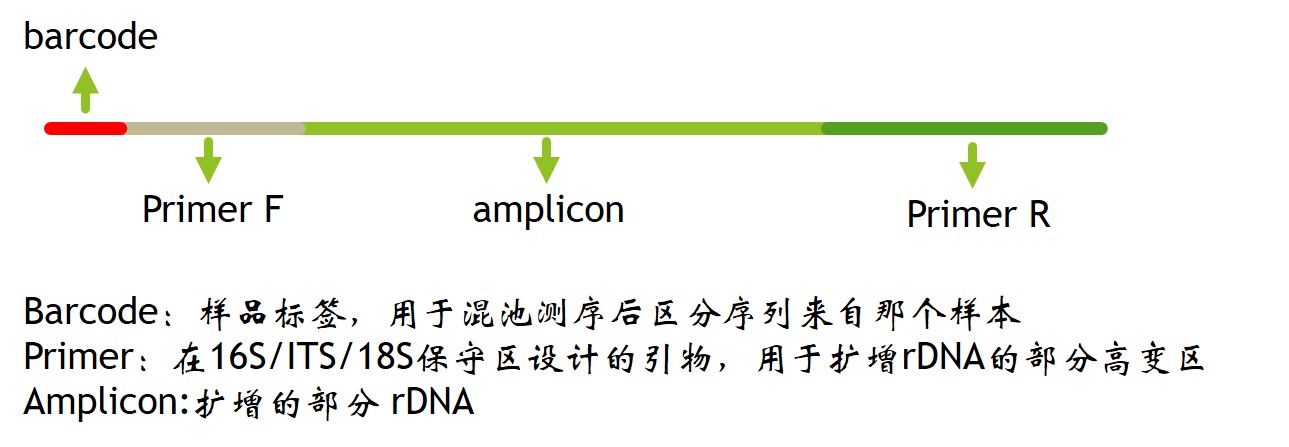
调研(低深度测序)主要用于找到合理建库方法----框架(高深度测序)----精细测序,进行修补
应用:
个体重测序----精准医疗----突变
重测序数据分析:GATK:人群队列测----发现SNP并汇总----发现群体高发SNP
转录组测序:
1.Small RNA seq表达
2.RNA seq表达-:--->Bowtie(短片段RNA比genome)---->Tophat(找到splice junction)----->cufflinks(找到可变剪接)
Bowtie is an ultrafast, memory-efficient short read aligner geared toward quickly aligning large sets of short DNA sequences (reads) to large genomes.
TopHat is a fast splice junction mapper for RNA-Seq reads.
Cufflinks assembles transcripts, estimates their abundances, and tests for differential expression and regulation in RNA-Seq samples.
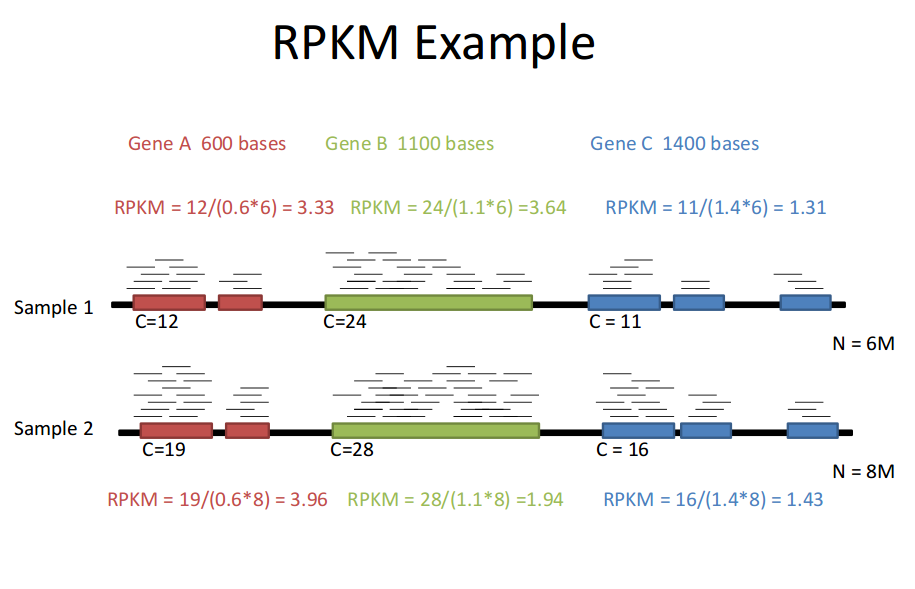
RPKM (Reads Per Kilobase of transcript per Million mapped reads)
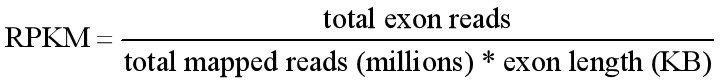
Total Exon reads:比到某个sample上的read数;
total mapped reads:比到某个sample上某gene exon的reads数
exon length:某gene exon长度
所以,
sample之间同一gene比较的是(gene mapped reads数/sample mapped reads数)
同一sample 的不同gene之间比较的是(gene mapped reads数/gene oxen length)
Chip-seq:发现转录因子的结合位点

