Fastqc使用说明
用FastQC检查二代测序原始数据的质量
2013-01-28 21:28:10| 分类: Bioinformatics | 标签:bioinformatics deep-seq |举报 |字号大中小 订阅
当二代测序的原始数据拿到手之后,第一步要做的就是看一看原始reads的质量。常用的工具就是fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)。fastqc的详细使用说明:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/
我们在服务器上用命令行来运行fastqc: fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN -o用来指定输出文件的所在目录,注意是不能自动新建目录的。输出的结果是.zip文件,默认自动解压缩,命令里加上--noextract则不解压缩。-f用来强制指定输入文件格式,默认会自动检测。-c用来指定一个contaminant文件,fastqc会把overrepresented sequences往这个 contaminant文件里搜索。contaminant文件的格式是"Name\tSequences",#开头的行是注释。加上 -q 会进入沉默模式,即不出现下面的提示: Started analysis of target.fq Approx 5% complete for target.fq Approx 10% complete for target.fq 如果输入的fastq文件名是target.fq,fastqc的输出的压缩文件将是target.fq_fastqc.zip。解压后,查看html格式的结果报告。结果分为如下几项:![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 结果分为绿色的"PASS",黄色的"WARN"和红色的"FAIL"。“You should treat the summary evaluations therefore as pointers to where you should concentrate your attention and understand why your library may not look random and diverse. ”
结果分为绿色的"PASS",黄色的"WARN"和红色的"FAIL"。“You should treat the summary evaluations therefore as pointers to where you should concentrate your attention and understand why your library may not look random and diverse. ”
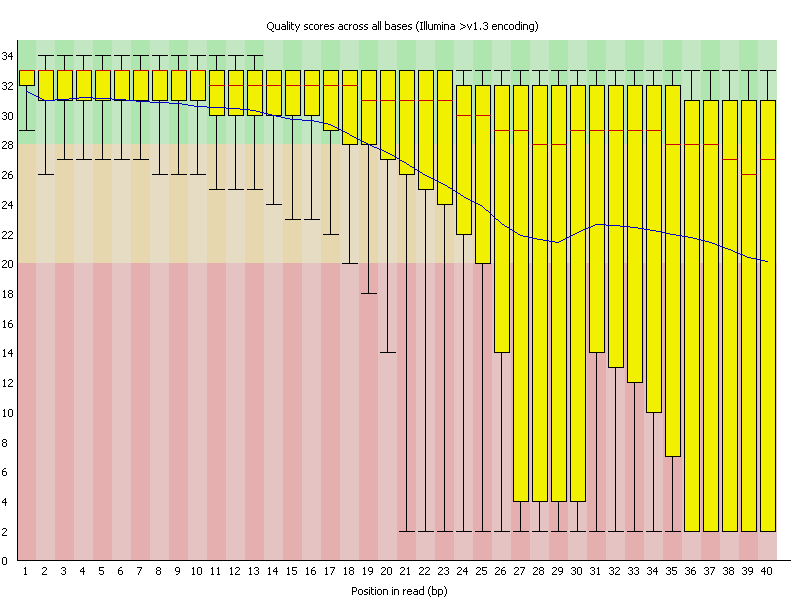
1 Basic statistics 如下面例子所示:![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 2 Per base sequence quality quality就是Fred值,-10*log10(p),p为测错的概率。所以一条reads某位置出错概率为0.01时,其quality就是20。图像如下面例子:
2 Per base sequence quality quality就是Fred值,-10*log10(p),p为测错的概率。所以一条reads某位置出错概率为0.01时,其quality就是20。图像如下面例子:
![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 横轴代表位置,纵轴quality。红色表示中位数,黄色是25%-75%区间,触须是10%-90%区间,蓝线是平均数。 若任一位置的下四分位数低于10或中位数低于25,报"WARN";若任一位置的下四分位数低于5或中位数低于20,报"FAIL".
横轴代表位置,纵轴quality。红色表示中位数,黄色是25%-75%区间,触须是10%-90%区间,蓝线是平均数。 若任一位置的下四分位数低于10或中位数低于25,报"WARN";若任一位置的下四分位数低于5或中位数低于20,报"FAIL".
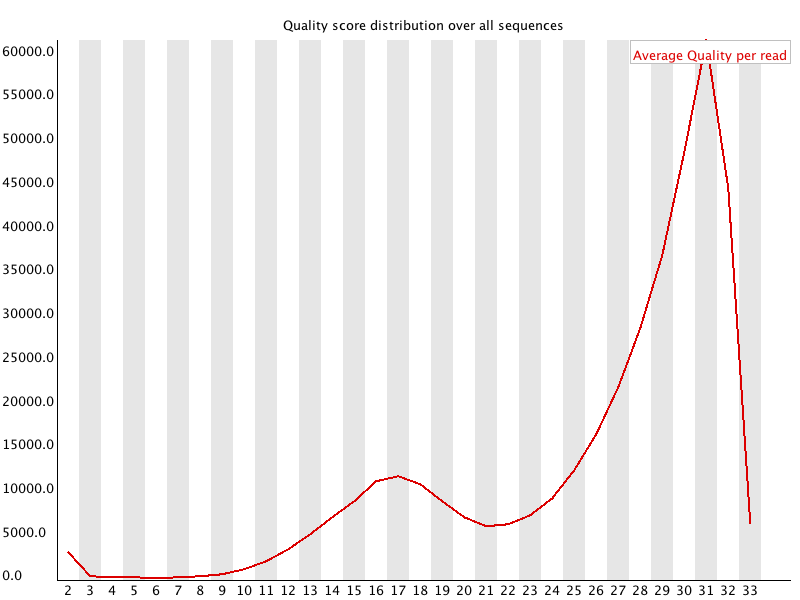
3 Per Sequence Quality Scores 每条reads的quality的均值的分布:![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 横轴为quality,纵轴是reads数目。当出现上图的情况时,我们就会知道有一部分reads具有比较差的质量。 当峰值小于27(错误率0.2%)时报"WARN",当峰值小于20(错误率1%)时报"FAIL"。
横轴为quality,纵轴是reads数目。当出现上图的情况时,我们就会知道有一部分reads具有比较差的质量。 当峰值小于27(错误率0.2%)时报"WARN",当峰值小于20(错误率1%)时报"FAIL"。
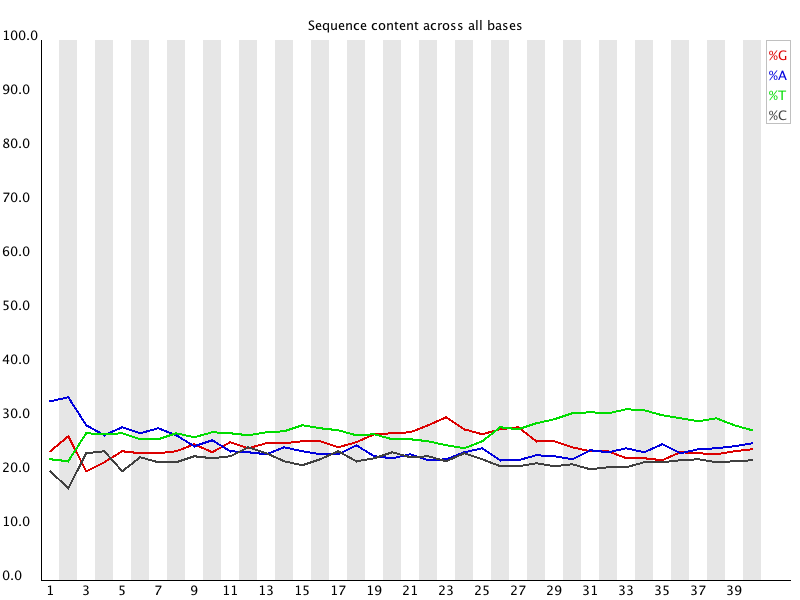
4 Per Base Sequence Content 对所有reads的每一个位置,统计ATCG四种碱基(正常情况)的分布:![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 横轴为位置,纵轴为百分比。 正常情况下四种碱基的出现频率应该是接近的,而且没有位置差异。因此好的样本中四条线应该平行且接近。当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。当所有位置的碱基比例一致的表现出bias时,即四条线平行但分开,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。 当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
横轴为位置,纵轴为百分比。 正常情况下四种碱基的出现频率应该是接近的,而且没有位置差异。因此好的样本中四条线应该平行且接近。当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。当所有位置的碱基比例一致的表现出bias时,即四条线平行但分开,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。 当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
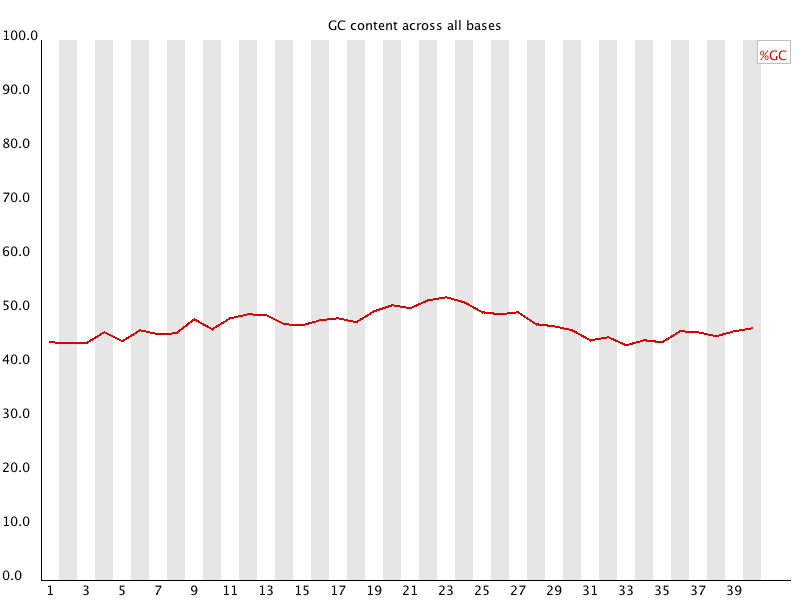
5 5 Per Base GC Content 对所有reads的每个位置,统计GC含量。![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 如果建库足够均匀,reads的每个位置应当是没有差异的,所以GC含量的线应当平行于X轴,反映样品(基因组、转录组等)的GC含量。当部分位置GC含量出现bias时,往往提示我们有overrepresented sequence的污染。当所有位置的GC含量一致的表现出bias时,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。 当任一位置的GC含量偏离均值的5%时,报"WARN";当任一位置的GC含量偏离均值的10%时,报"FAIL"。
如果建库足够均匀,reads的每个位置应当是没有差异的,所以GC含量的线应当平行于X轴,反映样品(基因组、转录组等)的GC含量。当部分位置GC含量出现bias时,往往提示我们有overrepresented sequence的污染。当所有位置的GC含量一致的表现出bias时,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。 当任一位置的GC含量偏离均值的5%时,报"WARN";当任一位置的GC含量偏离均值的10%时,报"FAIL"。
6 Per Sequence GC Content 统计reads的平均GC含量的分布。![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的)。 曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。 偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"。
红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的)。 曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。 偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"。
7 Per Base N Content 当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”。对所有reads的每个位置,统计N的比率:![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 正常情况下N的比例是很小的,所以图上常常看到一条直线,但放大Y轴之后会发现还是有N的存在,这不算问题。当Y轴在0%-100%的范围内也能看到“鼓包”时,说明测序系统出了问题。当任意位置的N的比例超过5%,报"WARN";当任意位置的N的比例超过20%,报"FAIL"。
正常情况下N的比例是很小的,所以图上常常看到一条直线,但放大Y轴之后会发现还是有N的存在,这不算问题。当Y轴在0%-100%的范围内也能看到“鼓包”时,说明测序系统出了问题。当任意位置的N的比例超过5%,报"WARN";当任意位置的N的比例超过20%,报"FAIL"。
8 Sequence Length Distribution reads长度的分布。![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 当reads长度不一致时报"WARN";当有长度为0的read时报“FAIL”。
当reads长度不一致时报"WARN";当有长度为0的read时报“FAIL”。
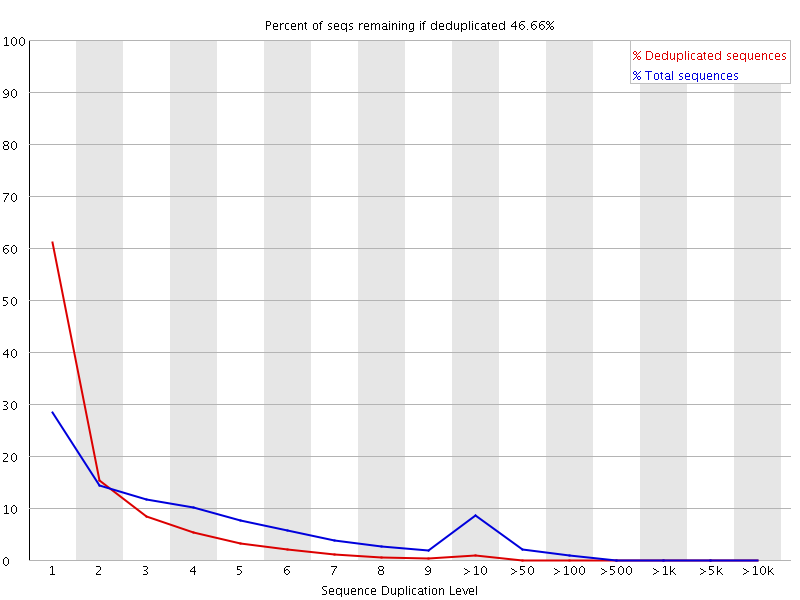
9 Duplicate Sequences 统计序列完全一样的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在(如建库过程中的PCR duplication)。![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 横坐标是duplication的次数,纵坐标是duplicated reads的数目,以unique reads的总数作为100%。 上图的情况中,相当于unique reads数目~20%的reads是观察到两个重复的,~7%是观察到三次重复的,依此类推。 可以想象,如果原始数据很大(事实往往如此),做这样的统计将非常慢,所以fastqc中用fq数据的前200,000条reads统计其在全部数据中的重复情况。重复数目大于等于10的reads被合并统计,这也是为什么我们看到上图的最右侧略有上扬。大于75bp的reads只取50bp(不知道怎么选的)进行比较。但由于reads越长越不容易完全相同(由测序错误导致),所以其重复程度仍有可能被低估。 当非unique的reads占总数的比例大于20%时,报"WARN";当非unique的reads占总数的比例大于50%时,报"FAIL“。
横坐标是duplication的次数,纵坐标是duplicated reads的数目,以unique reads的总数作为100%。 上图的情况中,相当于unique reads数目~20%的reads是观察到两个重复的,~7%是观察到三次重复的,依此类推。 可以想象,如果原始数据很大(事实往往如此),做这样的统计将非常慢,所以fastqc中用fq数据的前200,000条reads统计其在全部数据中的重复情况。重复数目大于等于10的reads被合并统计,这也是为什么我们看到上图的最右侧略有上扬。大于75bp的reads只取50bp(不知道怎么选的)进行比较。但由于reads越长越不容易完全相同(由测序错误导致),所以其重复程度仍有可能被低估。 当非unique的reads占总数的比例大于20%时,报"WARN";当非unique的reads占总数的比例大于50%时,报"FAIL“。
10 Overrepresented Sequences 如果有某个序列大量出现,就叫做over-represented。fastqc的标准是占全部reads的0.1%以上。和上面的duplicate analysis一样,为了计算方便,只取了fq数据的前200,000条reads进行统计,所以有可能over-represented reads不在里面。而且大于75bp的reads也是只取50bp。如果命令行中加入了-c contaminant file,出现的over-represented sequence会从contaminant_file里面找匹配的hit(至少20bp且最多一个mismatch),可以给我们一些线索。 当发现超过总reads数0.1%的reads时报”WARN“,当发现超过总reads数1%的reads时报”FAIL“。
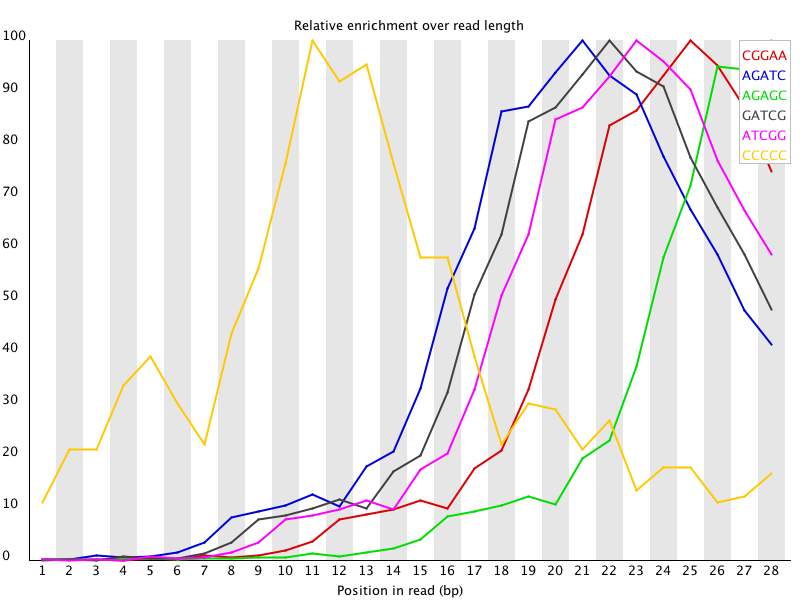
11 Overrepresented Kmers 如果某k个bp的短序列在reads中大量出现,其频率高于统计期望的话,fastqc将其记为over-represented k-mer。默认的k = 5,可以用-k --kmers选项来调节,范围是2-10。出现频率总体上3倍于期望或是在某位置上5倍于期望的k-mer被认为是over-represented。fastqc除了列出所有over-represented k-mers,还会把前6个的per base distribution画出来。![用FastQC检查二代测序原始数据的质量 - 静渊 - 静渊的学习日志]() 当有出现频率总体上3倍于期望或是在某位置上5倍于期望的k-mer时,报”WARN“;当有出现频率在某位置上10倍于期望的k-mer时报"FAIL"。
当有出现频率总体上3倍于期望或是在某位置上5倍于期望的k-mer时,报”WARN“;当有出现频率在某位置上10倍于期望的k-mer时报"FAIL"。
我们在服务器上用命令行来运行fastqc: fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN -o用来指定输出文件的所在目录,注意是不能自动新建目录的。输出的结果是.zip文件,默认自动解压缩,命令里加上--noextract则不解压缩。-f用来强制指定输入文件格式,默认会自动检测。-c用来指定一个contaminant文件,fastqc会把overrepresented sequences往这个 contaminant文件里搜索。contaminant文件的格式是"Name\tSequences",#开头的行是注释。加上 -q 会进入沉默模式,即不出现下面的提示: Started analysis of target.fq Approx 5% complete for target.fq Approx 10% complete for target.fq 如果输入的fastq文件名是target.fq,fastqc的输出的压缩文件将是target.fq_fastqc.zip。解压后,查看html格式的结果报告。结果分为如下几项:
 结果分为绿色的"PASS",黄色的"WARN"和红色的"FAIL"。“You should treat the summary evaluations therefore as pointers to where you should concentrate your attention and understand why your library may not look random and diverse. ”
结果分为绿色的"PASS",黄色的"WARN"和红色的"FAIL"。“You should treat the summary evaluations therefore as pointers to where you should concentrate your attention and understand why your library may not look random and diverse. ”1 Basic statistics 如下面例子所示:

3 Per Sequence Quality Scores 每条reads的quality的均值的分布:
4 Per Base Sequence Content 对所有reads的每一个位置,统计ATCG四种碱基(正常情况)的分布:
5 5 Per Base GC Content 对所有reads的每个位置,统计GC含量。
6 Per Sequence GC Content 统计reads的平均GC含量的分布。

7 Per Base N Content 当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”。对所有reads的每个位置,统计N的比率:

8 Sequence Length Distribution reads长度的分布。

9 Duplicate Sequences 统计序列完全一样的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在(如建库过程中的PCR duplication)。
10 Overrepresented Sequences 如果有某个序列大量出现,就叫做over-represented。fastqc的标准是占全部reads的0.1%以上。和上面的duplicate analysis一样,为了计算方便,只取了fq数据的前200,000条reads进行统计,所以有可能over-represented reads不在里面。而且大于75bp的reads也是只取50bp。如果命令行中加入了-c contaminant file,出现的over-represented sequence会从contaminant_file里面找匹配的hit(至少20bp且最多一个mismatch),可以给我们一些线索。 当发现超过总reads数0.1%的reads时报”WARN“,当发现超过总reads数1%的reads时报”FAIL“。
11 Overrepresented Kmers 如果某k个bp的短序列在reads中大量出现,其频率高于统计期望的话,fastqc将其记为over-represented k-mer。默认的k = 5,可以用-k --kmers选项来调节,范围是2-10。出现频率总体上3倍于期望或是在某位置上5倍于期望的k-mer被认为是over-represented。fastqc除了列出所有over-represented k-mers,还会把前6个的per base distribution画出来。
参考:http://www.plob.org/article/5987.html
http://yanshouyu.blog.163.com/blog/static/214283182201302835744453/

 浙公网安备 33010602011771号
浙公网安备 33010602011771号