GO分析-GOseq的使用教程
GOseq的介绍
GOseq是一个R包,用于寻找GO terms,即基因富集分析。此方法基于 Wallenius non-central hyper-geometric distribution。相对于普通的超几何分布(Hyper-geometric distribution),此分布的特点是从某个类别中抽取个体的概率与从某个类别之外抽取一个个体的概率是不同的,这种概率的不同是通过对基因长度的偏好性进行估计得到的,从而能更为准确地计算出 GO term 被差异基因富集的概率。
1.GOseq的安装
>BiocManager::install("goseq")
2.参考数据集
这里我们采用GOseq包里的内置数据集genes来做GO分析
1 library(goseq) 2 data(genes) 3 head(genes) 4 str(genes)
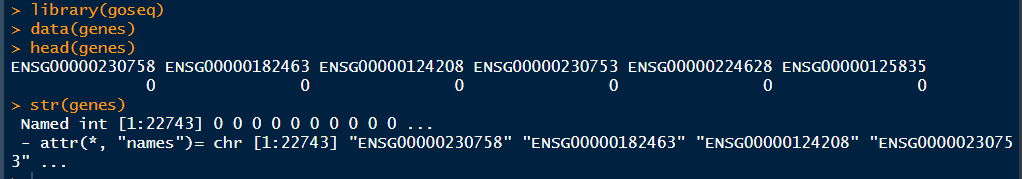
这里genes数据集是EMSEMBL gene的向量集合,其中1代表差异表达
3.通过getgo函数获得GO terms
getgo的用法:
1 getgo(genes, genome, id,fetch.cats=c("GO:CC","GO:BP","GO:MF"))
genes:genes是输入的gene向量或列表
genome:参考基因组,比如hg38,hg19
id:输入基因的类型,比如ensGene
fetch,cats:fetch.cats是"GO:CC", "GO:BP", "GO:MF" & "KEGG"的一系列组合
这里用supportedOrganisms()函数来查看支持的genome和id
结果:结果是一个列表,包含每一个gene对应的所有的GO ID,这个值是goseq函数中gene2cat参数的输入值
举例:
1 genes <- c("ENSG00000124208", "ENSG00000182463", "ENSG00000124201", "ENSG00000124205", "ENSG00000124207") 2 getgo(genes,'hg19','ensGene')
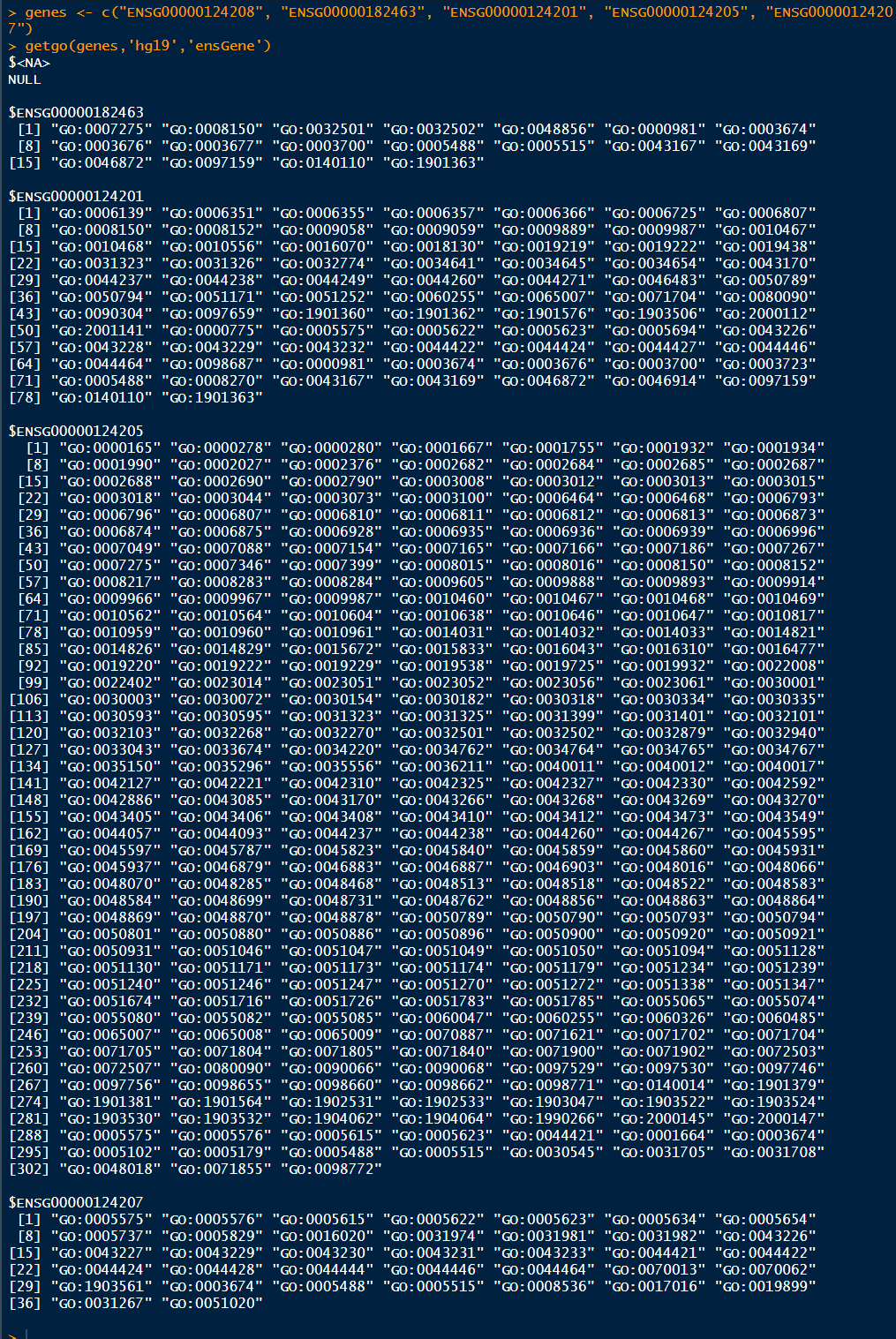
这里显示了每一个gene参与到的所有GO ID
3.通过getlength函数检索gene的长度
getlength用法:
1 getlength(genes, genome, id)
结果:结果是一个向量,包含所有基因的长度,如果某个基因的长度无法检索到,用NA代替。这个向量是nullp函数中bias.data的输入值。
举例:
1 genes <- c("ENSG00000124208", "ENSG00000182463", "ENSG00000124201", "ENSG00000124205", "ENSG00000124207") 2 getlength(genes,'hg19','ensGene')
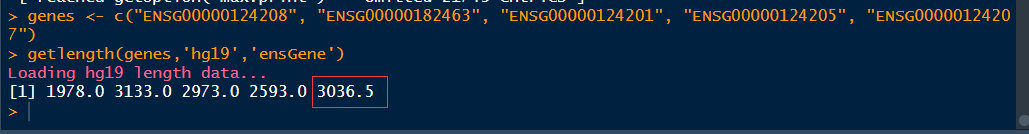
这里基因长度出现了3036.5,是因为这里基因长度取得是转录本长度的中位数。
4.使用nullp函数(Probability Weighting Function)计算概率加权函数
nullp函数介绍:
Calculates a Probability Weighting Function for a set of genes based on a given set of biased data (usually gene length) and each genes status as differentially expressed or not.
nullp函数用法:
1 nullp(DEgenes, genome, id, bias.data=NULL,plot.fit=TRUE)
DEgenes:DEgenes的格式是一个二元向量,其中1代表差异表达,0代表非差异表达,还有包括gene id,格式与内置数据集genes一样
bias.data:bias.data是一个数值向量,通常是基因转录本长度的中位数,单位是bp.如果设置bias.data=NULL,nullp函数将通过getlength函数来获取gene的长度。所以这里默认设置为bias.data=NULL
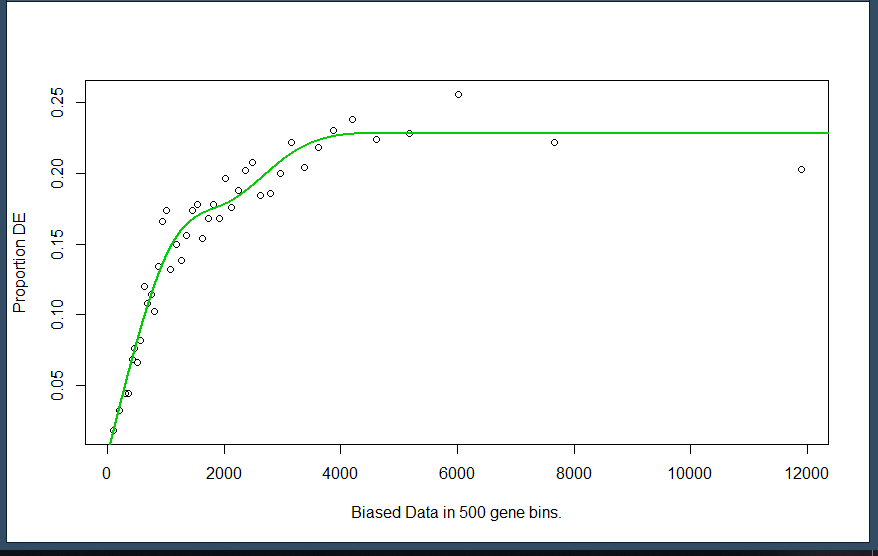
plot.fit:plot.fit这里将pwf作图,默认设置为plot,fit=TRUE
一般nullp函数后面的参数都选择默认,只用选择设置DEgenes, genome和 id即可
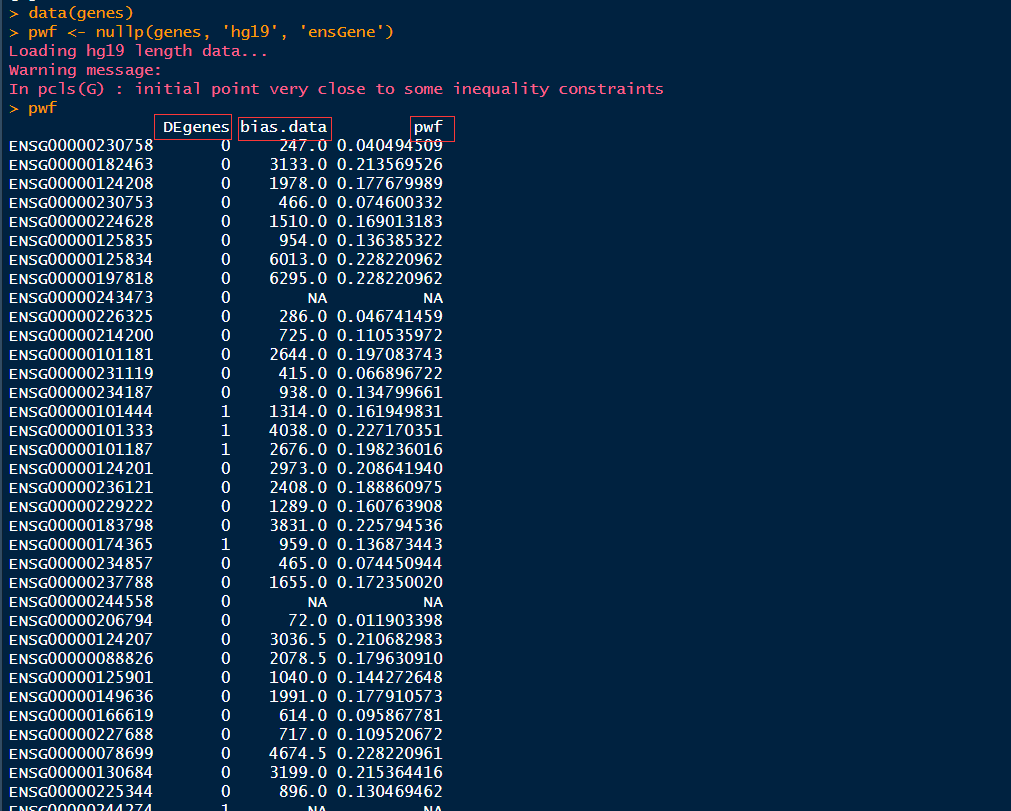
结果:结果是一个数据框,行名为gene id,列名为"DEgenes", "bias.data" 和 "pwf",这个数据框对象是goseq函数的输入,用来计算富集的GO terms,也可以作为plotPWF的输入,用来进一步作图。
举例:
1 data(genes) 2 pwf <- nullp(genes, 'hg19', 'ensGene')
5.使用goseq函数进行GO富集分析
goseq函数介绍:
Does selection-unbiased testing for category enrichment amongst differentially expressed (DE) genes for RNA-seq data. By default, tests gene ontology (GO) categories, but any categories may be tested.
goseq函数用法:
1 goseq(pwf, genome, id, gene2cat = NULL,test.cats=c("GO:CC", "GO:BP", "GO:MF"),method = "Wallenius", repcnt = 2000, use_genes_without_cat=FALSE)
pwf:这里的pwf是由nullp函数得到的结果,为一个数据框
gene2cat:这里的gene2cat是由getgo函数得到的结果,如果设置gene2cat=NULL,goseq函数将会自动地用getgo函数来获得GO ID,默认设置是gene2cat=NULL
method:这里method有三种选择,"Wallenius", "Sampling" 和 "Hypergeometric".这里"Sampling" 和 "Hypergeometric"方法几乎从没被使用过
一般goseq函数后面的参数都可以选择默认,只用选择pwf,genome和id这三个参数就可以
举例:
1 data(genes) 2 pwf <- nullp(genes,'hg19','ensGene') 3 pvals <- goseq(pwf,'hg19','ensGene') 4 head(pvals)
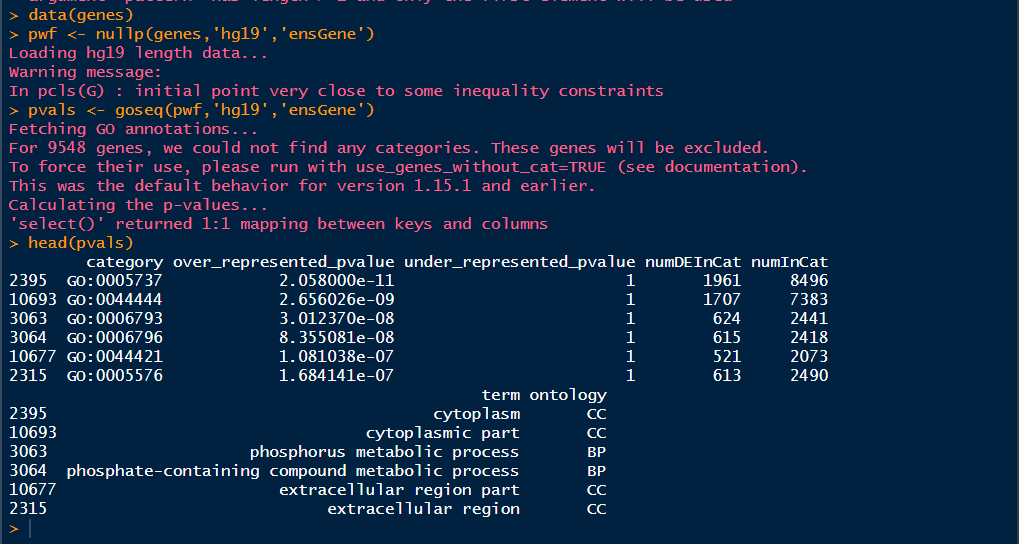
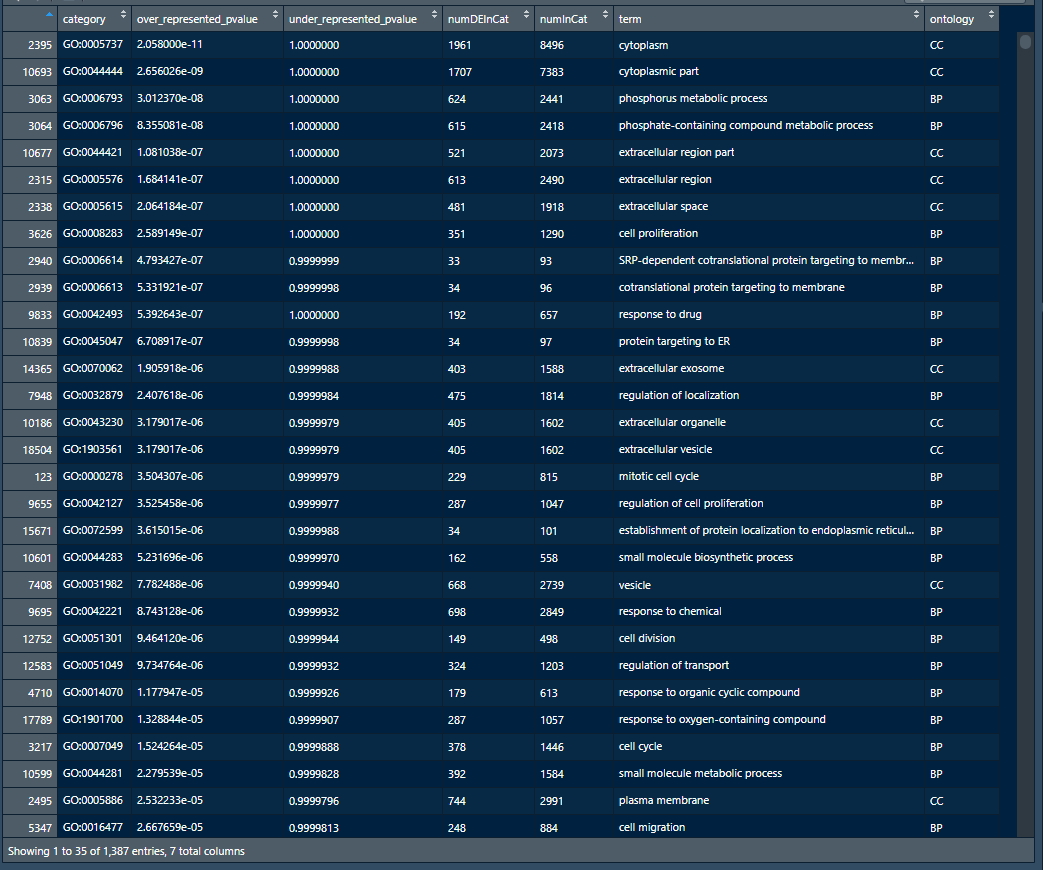
这里的选择over_represented_pvalues<0.05就是具有统计学意义的GO ID了
1 enriched.GO<-pvals[pvals$over_represented_pvalue<0.05,]