
bioMart进行基因id的转换
1.bioMart包的介绍
bioMart包是一个连接bioMart数据库的R语言接口,能通过这个软件包自由连接到bioMart数据库,这个包可以做以下几个工作:
1.查找某个基因在染色体上的位置。反之,给定染色体每一区间,返回该区间的基因;
2.通过EntrezGene的ID查找到相关序列的GO注释。反之,给定相关的GO注释,获取相关的EntrezGene的ID;
3.通过EntrezGene的ID查找到相关序列的上游100bp序列(可能包含启动子等调控元件);
4.查找人类染色体上每一段区域中已知的SNPs;
5.给定一组的序列ID,获得其中具体的序列;
2.bioMart的安装以及基本功能
1.biomart的安装biomaRt需要通过biomanager来安装,首先安装biomanager,然后安装biomaRt
> install.packages("BiocManager") > BiocManager::install("biomaRt")
2.载入biomaRt并显示一下能连接的数据库
>library("biomaRt") >listMarts()
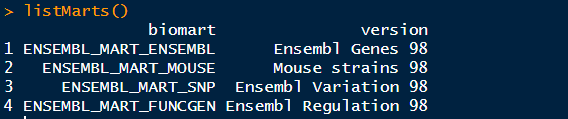
这里我们选择ensembl数据库
3..用useMart函数选定数据库
>mart<-useMart("emsembl")
这里我们要获取的基因注释的基因是人类基因,所以选择hsapiens_gene_ensembl,通过以下代码来选择emsembl库的人类参考基因组来分析
mart <- useMart("ensembl","hsapiens_gene_ensembl")##小鼠选择mmusculus_gene_ensembl
4.用listFilters()函数选定输入的ID类型
然后,你需要知道输入的是什么类型的ID,即转换前的ID类型,如:
ENSG00000000003或ENMUSG000000003,属于类型为ensembl_gene_id;
ENST00000000233或ENMUST00000000233,属于类型为ensembl_transcript_id;
102178245,属于类型为entrezgene;
Hoxc13,属于类型为external_gene_name;
NM_000014,属于类型为refseq_mrna;
hsa-let-7a-1,属于类型为mirbase_id;
用listFilters()函数查看可选择的输入类型,listFilters(mart)一共显示了446行,一共包括name和description两列,这里只截取一部分
>listFilters(mart)

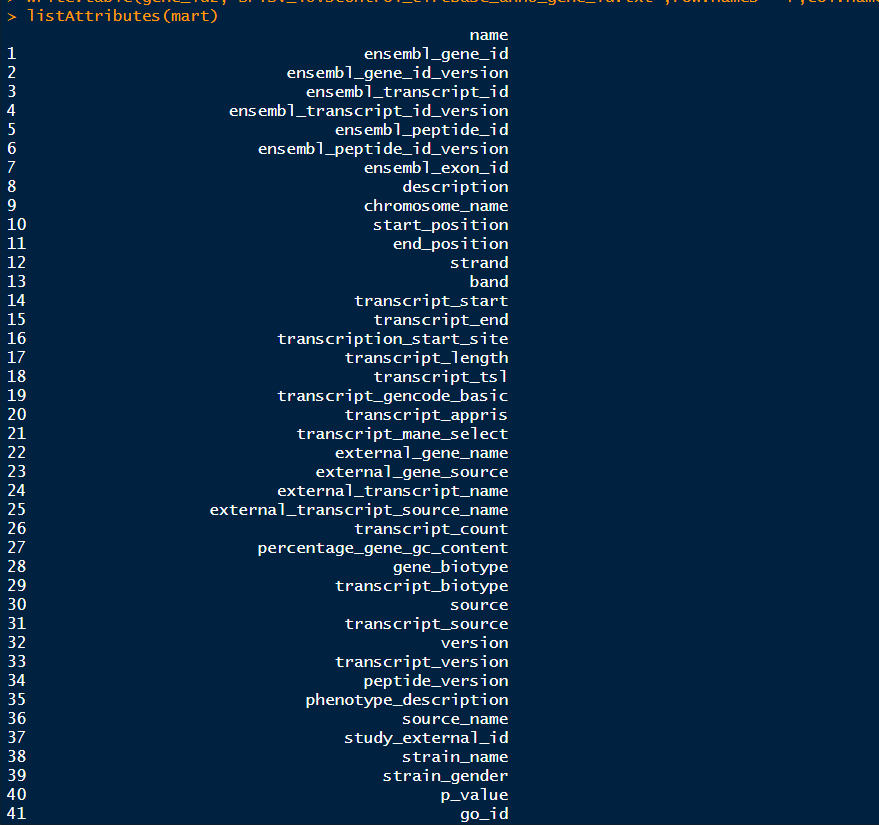
5.用listAttributes()函数选定输出的ID类型
要想知道biomaRt支持哪些ID类型的输出,可以通过以下命令查看,共支持3607种ID输出,这里只截取了一部分输出,一共有name和description两列
listAttributes(mart)
6.用getBM()函数获取注释
hg_symbols<- getBM(attributes=c('ensembl_gene_id','hgnc_symbol',"chromosome_name", "start_position","end_position", "band"), filters= 'ensembl_gene_id', gene = my_ensembl_gene_id, mart = mart)
这个函数有4个参数
filters()里面的值为我们输入的ID类型
gene= 这个值就是我们要输入的数据
mart= 这个值是我们所选定的数据库的基因组
3.biomaRt实例操作
setwd("C:/Users/Desktop/circRNA分析结果/find_circ_result自己分析结果")#设置working directory gene_symbol<-read.csv("SFTSV_24vscontrol_circBase_anno.csv",header=F,stringsAsFactors = F)[,11]#读取数据并提取含有gene_symbol的列 library(biomaRt) mart <- useMart("ensembl","hsapiens_gene_ensembl")##小鼠选择mmusculus_gene_ensembl gene_id<-getBM(attributes=c("external_gene_name","ensembl_gene_id"),filters = "external_gene_name",values = gene_symbol, mart = mart)#将输入的filters设置未external_gene_name(也就是gene_symbol),将输出的attributes设置为external_gene_name和emsembl_gene_id write.table(gene_id,"SFTSV_24vscontrol_circBase_anno_gene_id.txt",row.names = F,col.names=F,quote=F)

