
测序原理
1. 高通量测序(highthroughput sequencing, HTS)/下一代测序(next generation sequencing, NGS)前世今生
ref: https://baijiahao.baidu.com/s?id=1612934763948082306&wfr=spider&for=pc
共有三代HTS方法,
第一代:Sanger测序(已淘汰)
双脱氧核苷酸末端终止法。引物结合模板/sample, DNsae延伸引物,掺入ddNTP在每一个base位置终止链反应,拼接不同长度合成序列得到所有序列
缺点:慢
衍生:鸟枪法。打断长DNA成若干短片段,分别对每个短片段sanger测序从而大大节约时间
第二代:Roche 454焦磷酸测序,Illumina Solexa测序,ABI SOLID测序 (目前主流)
目前主要使用Illumina Solexa测序。“sequencing by synthesis”通过合成进行测序(De novo sequencing)。先"加adapter使目的片段与flowcell连接->PCR扩增目的片段(fluo-tag dNTP)->酶消化反义链只留原始目的片段,得到荧光标记目的片段簇->detect fluorescence(i.e.sequencing)
第三代:单分子实时DNA测序(no PCR, 尚未广泛应用)
单分子荧光测序(边合成边测序);纳米孔测序(电信号)
2.单端测序(Single-read)和双端测序(Paired-end和Mate-pair)的关系
ref:https://blog.csdn.net/hanli1992/article/details/82982434
main difference : construct sequencing library(接adapter方法不同)
Single end:
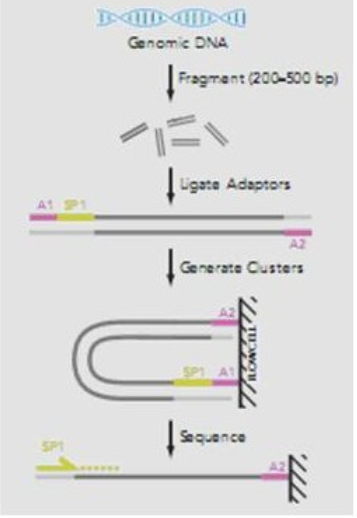
碎片化DNA->加sequencing primer(SP1)至片段一端后,两端加adapter(A1,A2)->利用adapter将碎片化目的片段固定于flowcell生成DNA簇(2链由于PCR与1链配对故而扩增的2链有SP1识别位点)->由于SP1测序方向的设计,只有2链会被sequencing而1链不会被测序
“adapter有一对但SP只有一个,一次测序”
pair end:
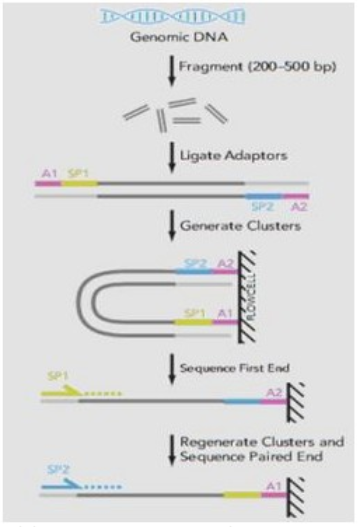
与SE类似,但“adapter和SP都有一对,分别连在DNA两条链两个方向上,两次测序”
先测2链->去除2链->用对读测序模块(Paired-End Module)引导2链互补链在原位置再生和扩增形成1链DNA簇->1链测序
3.call mutation去重
mutation来源:cell本身(我们真正想要得到的真实mutation)+ 碎片化DNA时引入error1 + PCR扩增error2
细胞量不足或者提取DNA时发生降解都可能造成局部DNA浓度低,直接测序有可能损失该部分微量DNA,故而使用PCR扩增。
PCR扩增产生的重复序列可能造成mutation calling 的不准确:
(1)假阳性变异:原本量很多的DNA通过PCR更多了,故取样测序时可能被重复取样从而产生测序重复序列,这样error1&error2都被放大
(2)假阴性变异:由于PCR Bias(PCR可能倾向于更多扩增含有某一碱基的DNA片段),若这个片段恰巧是call mutation用的reference片段上的碱基,那么reference信号的增强也意味着mutation信号的减弱,从而被误判为不重要的mutation
GATK、Samtools、Platpus等这种利用贝叶斯原理的变异检测算法都是认为所用的序列数据都不是重复序列(即将它们和其他序列一视同仁地进行变异的判断,所以带来误导),因此必须要进行标记(去除)或者使用PCR-Free的测序方案(这种方案目前正变得越来越流行,特别是对于RNA-Seq来说尤为重要,现在著名的基因组学研究所——Broad Institute,基本都是使用PCR-Free的测序方案)
这也提醒我们用这些软件时要记得remove duplicate

