

2022-006 在bam中检查指定突变
转载
2022-006 在bam中检查指定突变

需求
检查突变在bam文件中存不存在。
注意:以下操作均需要bam文件按坐标排序并建立索引。
$ samtools sort -@ 24 -o sorted.bam un_sorted.bam
$ samtools index sorted.bam参考基因组经常用到,因此赋值到变量。
hg19=~/project/0-reference/2-GATK/hg19/human_g1k_v37_decoy.fasta示例突变为一个SNP和一个INDEL:
- SNP 10_100021916_T_C 10号染色体100021916处T到C的单碱基突变
- INDEL 10_102775349_CT_C 10号染色体102775349处CT到C的1bp删除
方案一:直接看
使用samtools命令可以对bam文件的指定位置进行展示。
10_100021916_T_C:
$ samtools tview -p 10:100021916 test1.bam ${hg19}
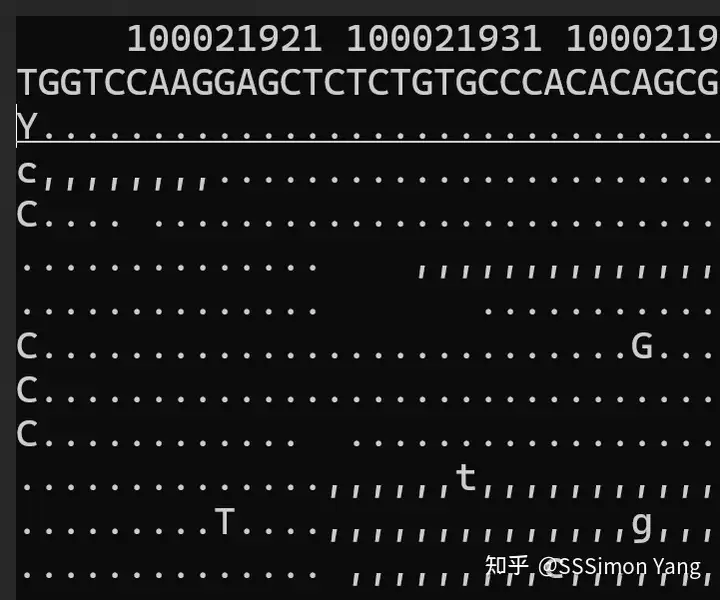
有多条reads在T位置为C,表示突变存在。
10_102775349_CT_C:
$ samtools tview -p 10:102775349 test2.bam ${hg19}
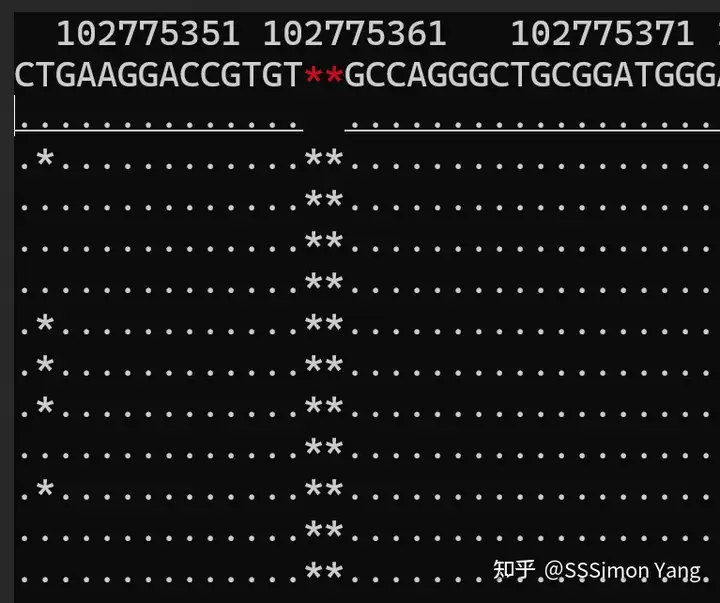
有多条reads在C后的T位置为*,表缺失,表示突变存在。
也可以使用IGV (Integrative Genomics Viewer) 对指定区域的reads进行可视化。
10_100021916_T_C:
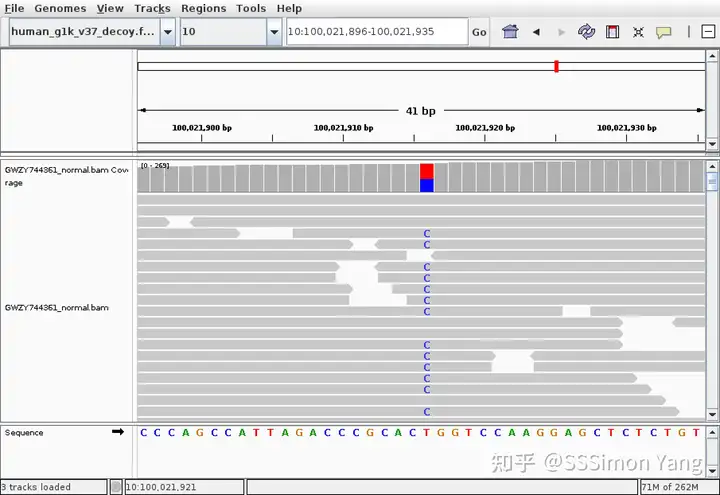
10_102775349_CT_C:
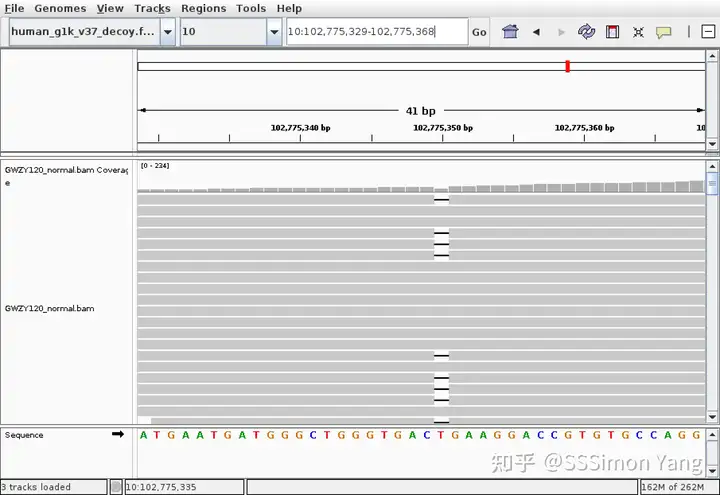
与samtools相比,IGV可以更清楚地可视化duplicate reads等信息,但在操作上略麻烦。
方案优点:简单,能够直接观察reads的质量。
方案缺点:每次只能检查一个bam,不能根据阈值对reads进行筛选。
方案二:输出碱基数
使用sambamba,可以输出指定位置的碱基数。
10_100021916_T_C:
$ sambamba depth base -F '' -L 10:100021916-100021916 test1.bam
REF POS COV A C G T DEL REFSKIP SAMPLE
Processing reference #10 (10)
10 100021915 306 0 140 0 166 0 0 test1可以看到C有140个,T有166个,表示突变存在。
10_102775349_CT_C:
$ sambamba depth base -F '' -L 10:102775349-102775350 test2.bam
REF POS COV A C G T DEL REFSKIP SAMPLE
Processing reference #10 (10)
10 102775348 44 0 44 0 0 0 0 test2
10 102775349 48 0 2 0 26 20 0 test2可以看到第二行的T有26个,DEL有20个,表示突变存在。
sambamba的输出是0-based,所以坐标会自动减一位。
在命令中的-F ''可以替换成其他的筛选条件,其默认值为mapping_quality > 0 and not duplicate and not failed_quality_control。详见filter expression syntax。
sambamba可以同时检查多个bam。
$ sambamba depth base -F '' -L 10:100021916-100021916 *.bam
REF POS COV A C G T DEL REFSKIP SAMPLE
Processing reference #10 (10)
10 100021915 306 0 140 0 166 0 0 test1
10 100021915 352 0 0 0 352 0 0 test2对输出文件稍加处理即可确定突变的存在与否。
方案优点:能够对reads进行阈值筛选,能够同时处理多个bam。
方案缺点:对INDEL的处理较为麻烦。
方案三:指定位置call突变
call突变有很多工具,最简单的工具就是bcftools。而bcftools恰好能够只在指定位置call突变。
10_100021916_T_C:
$ bcftools mpileup -r 10:100021916 -f ${hg19} -Ou test1.bam | bcftools call -mv
...
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT test1
10 100021916 . T C 222.385 . DP=240;VDB=0.312239;SGB=-0.693147;RPBZ=-0.522758;BQBZ=2.65699;SCBZ=-1.51572;FS=0;MQ0F=0;AC=1;AN=2;DP4=41,25,34,26;MQ=60 GT:PL 0/1:255,0,255突变存在。
10_102775349_CT_C:
$ bcftools mpileup -r 10:102775349 -f ${hg19} -Ou test2.bam | bcftools call -mv
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT test2
10 102775349 . CT C 138.301 . INDEL;IDV=16;IMF=0.421053;DP=38;VDB=2.58825e-06;SGB=-0.689466;RPBZ=-0.222168;FS=0;MQ0F=0;AC=1;AN=2;DP4=15,7,14,2;MQ=60 GT:PL 0/1:171,0,143突变存在。
bcftools可以同时处理多bam的多区域。
$ cat test
10 100021916
10 102775349
$ bcftools mpileup -R test -f ${hg19} -q 10 -Q 10 --ff UNMAP -Ou *.bam | bcftools call -mv
...
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT test1 test2
10 100021916 . T C 221.283 . DP=544;VDB=0.206128;SGB=49.7367;RPBZ=-0.237584;BQBZ=-1.49091;SCBZ=-0.406369;FS=0;MQ0F=0;AC=1;AN=4;DP4=146,92,46,28;MQ=60 GT:PL 0/1:255,0,255 0/0:0,255,255
10 102775349 . CT C 150.169 . INDEL;IDV=20;IMF=0.454545;DP=82;VDB=1.39796e-09;SGB=12.3071;RPBZ=-0.746538;SCBZ=-0.567962;FS=0;MQ0F=0;AC=1;AN=4;DP4=45,14,17,3;MQ=60 GT:PL 0/0:0,105,194 0/1:184,0,143通过指定-q -Q --ff等参数对reads进行筛选。--ff的默认值是UNMAP,SECONDARY,QCFAIL,DUP。详见mpileup。
bcftools的输出为vcf文件,确定突变存在与否需要对其进行的文本处理更为简单。
方案优点:能够对reads进行阈值筛选,能够处理多个bam,能够比较好的处理INDEL。
END
总的来说,使用bcftools以call突变的方式检查指定突变的存在是目前最好的解决方案。我也尝试过使用gatk实现该需求,但是gatk并不能只在指定位置call突变,因此废弃。
我
我是 SSSimon Yang,关注我,用code解读世界
