GATK使用说明-GRCh38(Genome Reference Consortium)(二)
Reference Genome Components
1. GRCh38 is special because it has alternate contigs that represent population haplotypes.
Don’t know alternate contig from alternate dimension? Spend five minutes now to review terminology in our Dictionary entryReference Genome Components. At the least, you should understand the distinction between the primary assembly and alternate contigs.
Long BAM headers notwithstanding, GRCh38 alternate contig sequences are only ~3.6% of the primary assembly length (see table). They encompass alternate haplotypes for which we cannot easily represent variants on the primary assembly. According to my estimation, roughly a tenth of a percent (101,845 basepairs) of the alternate sequence appears highly divergent.
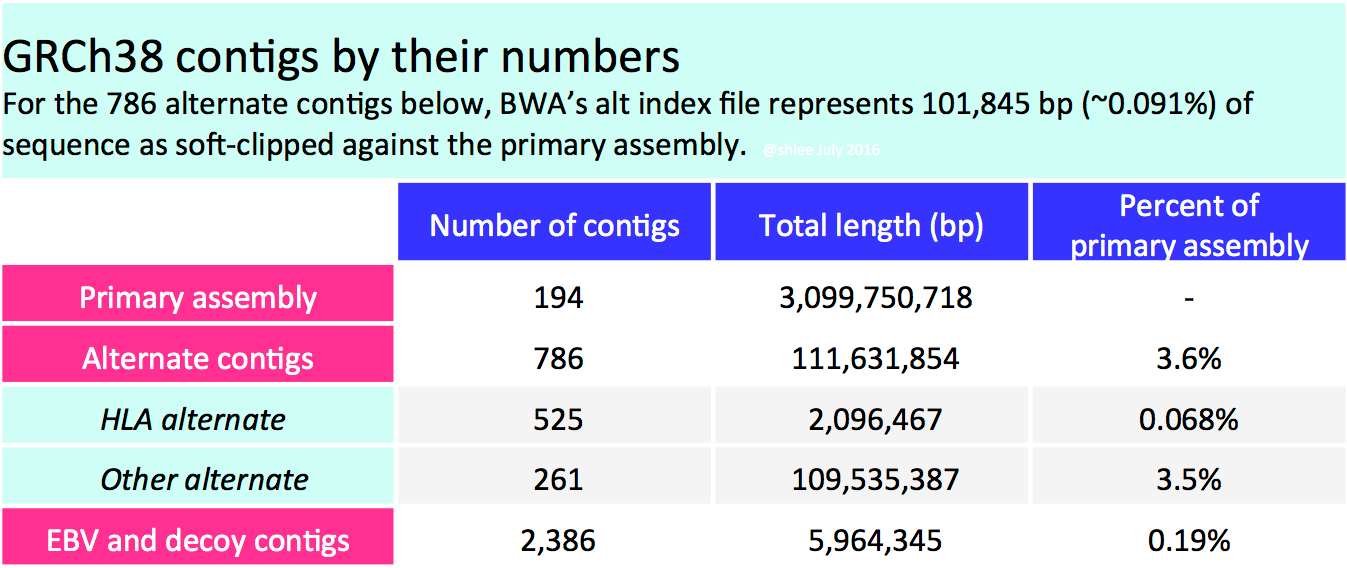
1.1 Reference Genome Components
This document defines several components of a reference genome. We use the human GRCh38/hg38 assembly to illustrate.
GRCh38/hg38 is the assembly of the human genome released December of 2013, that uses alternate or ALT contigs to represent common complex variation, including HLA loci. Alternate contigs are also present in past assemblies but not to the extent we see with GRCh38. Much of the improvements in GRCh38 are the result of other genome sequencing and analysis projects, including the 1000 Genomes Project.
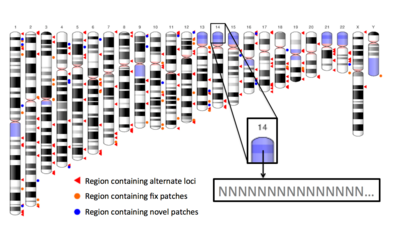
The ideogram is from the Genome Reference Consortium website and showcases GRCh38.p7. The zoomed region illustrates how regions in blue are full of Ns.
Analysis set reference genomes have special features to accommodate sequence read alignment. This type of genome reference can differ from the reference you use to browse the genome.
For example, the GRCh38 analysis set hard-masks, i.e. replaces with Ns, duplicate copies of centromeric着丝粒 and genomic repeat arrays (on chromosomes 5, 14, 19, 21, & 22,Satellite DNA consists of very large arrays of tandemly repeating, non-coding DNA. Satellite DNA is the main component of functional centromeres, and form the main structural constituent of heterochromatin.) and two PAR regions on chromosome Y. Confirm the set you are using by viewing a PAR region of the Y chromosome on IGV as shown in the figure below. The chrY location of PAR1 and PAR2 on GRCh38 are chrY:10,000-2,781,479 and chrY:56,887,902-57,217,415.

- The sequence in the reference set is a mix of uppercase and lowercase letters. The lowercase letters represent soft-masked sequence corresponding to repeats from RepeatMasker and Tandem Repeats Finder.
- The GRCh38 analysis sets also include a contig to siphon off reads corresponding to the Epstein-Barr virus sequence as well as decoy诱骗 contigs. The EBV contig can help correct for artifacts stemming from(起源于) immortalization无限增殖化 of human blood lymphocytes淋巴细胞 with EBV transformation转化, as well as capture endogenous内生的 EBV sequence as EBV naturally infects B cells in ~90% of the world population. Heng Li provides the decoy contigs.
1.2 Nomenclature: words to describe components of reference genomes
-
A contig is a contiguous sequence without gaps.
-
Alternate contigs, alternate scaffolds or alternate loci allow for representation of diverging haplotypes. These regions are too complex for a single representation. Identify ALT contigs by their
_altsuffix.The GRCh38 ALT contigs total 109Mb in length and span 60Mb of the primary assembly. Alternate contig sequences can be novel to highly diverged or nearly identical to corresponding primary assembly sequence. Sequences that are highly diverged from the primary assembly only contribute a few million bases. Most subsequences of ALT contigs are fairly similar to the primary assembly. This means that if we align sequence reads to GRCh38+ALT blindly, then we obtain many multi-mapping reads with zero mapping quality. Since many GATK tools have a ZeroMappingQuality filter, we will then miss variants corresponding to such loci.
-
Primary assembly refers to the collection of (i) assembled chromosomes, (ii) unlocalized and (iii) unplaced sequences. It represents a non-redundant haploid genome.
(i) Assembled chromosomes for hg38 are chromosomes 1–22 (
chr1–chr22), X (chrX), Y (chrY) and Mitochondrial (chrM). (ii)Unlocalized sequence are on a specific chromosome but with unknown order or orientation. Identify by_randomsuffix. (iii) Unplacedsequence are on an unknown chromosome. Identify bychrU_prefix. -
PAR stands for pseudoautosomal region. PAR regions in mammalian X and Y chromosomes allow for recombination between the sex chromosomes. Because the PAR sequences together create a diploid or pseudo-autosomal sequence region, the X and Y chromosome sequences are intentionally identical in the genome assembly. Analysis set genomes further hard-mask two of the Y chromosome PAR regions so as to allow mapping of reads solely to the X chromosome PAR regions.
- ---人类Y染色体属近端着丝粒染色体,由长臂Yq和微小的短臂Yp组成,DNA长度约60Mb。Y染色体两端各有一小部分称拟常染色区(pseudoautosomal region),在减数分裂过程中,拟常染区可与X染色体的相应区段进行交换、重组。
-
Different assemblies shift coordinates for loci and are released infrequently. Hg19 and hg38 represent two different major assemblies. Comparing data from different assemblies requires lift-over tools that adjust genomic coordinates to match loci, at times imperfectly. In the special case of hg19 and GRCh37, the primary assembly coordinates are identical for loci but patch updates differ. Also, the naming conventions of the references differ, e.g. the use of chr1 versus 1 to indicate chromosome 1, such that these also require lift-over to compare data. GRCh38/hg38 unifies the assemblies and the naming conventions.
-
Patches are regional fixes that are released periodically for a given assembly. GRCh38.p7 indicates the seventh patched minor release of GRCh38. This NCBI page explains in more detail. Patches add information to the assembly without disrupting the chromosome coordinates. Again, they improve representation without affecting chromosome coordinate stability. The two types of patches, fixed and novel, represent different types of sequence.
(i) Fix patches represent sequences that will replace primary assembly sequence in the next major assembly release. When interpreting data, fix patches should take precedence over the chromosomes. (ii) Novel patches represent alternate loci. When interpreting data, treat novel patches as population sequence variants.
1.3 The GATK perspective on reference genomes
Within GATK documentation, Tutorial#8017 outlines how to map reads in an alternate contig aware manner and discusses some of the implications影响 of mapping reads to reference genomes with alternate contigs.
GATK tools allow for use of a genomic intervals list that tells tools which regions of the genome the tools should act on. Judicious明智的 use of an intervals list, e.g. one that excludes排除 regions of Ns and low complexity repeat regions in the genome, makes processes more efficient. This brings us to the next point.
Specifying contigs with colons冒号 in their names, as occurs for new contigs in GRCh38, requires special handling操作 for GATK versions prior to v3.6. Please use the following workaround工作区.
- For example,
HLA-A*01:01:01:01is a new contig in GRCh38. The colons are a new feature of contig naming for GRCh38 from prior assemblies. This has implications暗示 for using the-Loption of GATK as the option also uses the colon as a delimiter分隔符 to distinguish between contig and genomic coordinates. - When defining coordinates of interest for a contig, e.g. positions 1-100 for chr1, we would use
-L chr1:1-100. This also works for our HLA contig, e.g.-L HLA-A*01:01:01:01:1-100. -
However, when passing in an entire整个 contig, for contigs with colons in the name, you must add
:1+to the end of the chromosome name as shown below. This ensures that portions of the contig name are appropriately合适地 identified识别 as part of the contig name and not genomic coordinates.-L HLA-A*01:01:01:01:1+
1.4 Viewing CRAM alignments on genome browsers
Because CRAM compression压缩 depends on the alignment reference genome, tools that use CRAM files ensure correct decompression解压 by comparing reference contig MD5 hashtag values. These are sensitive to any changes in the sequence, e.g. masking with Ns. This can have implications for viewing alignments in genome browsers when there is a disjoint(互斥,不相交) between the reference that is loaded in the browser and the reference that was used in alignment. If you are using a version of tools for which this is an issue, be sure to load the original analysis set reference genome to view the CRAM alignments.
1.5 Should I switch to a newer reference?
Yes you should. In addition to adding many alternate contigs, GRCh38 corrects thousands of SNPs and indels in the GRCh37 assembly that are absent in the population and are likely sequencing artifacts. It also includes synthetic合成的 centromeric着丝粒 sequence and updates non-nuclear genomic sequence.
The ability to recognize alternate haplotypes for loci is a drastic improvement that GRCh38 makes possible. Going forward, expanding genomics data will help identify variants for alternate haplotypes, improve existing and add additional alternate haplotypes and give us a better accounting of alternate haplotypes within populations. We are already seeing improvements and additions in the patch releases to reference genomes, e.g. the seven minor releases of GRCh38 available at the time of this writing.
Note that variants produced by alternate haplotypes when they are represented on the primary assembly may or may not be present in data resources, e.g. dbSNP. This could have varying degrees of impact, including negligible可以忽略, for any process that relies on known variant sites. Consider the impact this discrepant不一致的 coverage in data resources may have for your research aims and weigh this against the impact of missing variants because their sequence context is unaccounted for in previous assemblies.
2. The GRCh38 analysis set hard-masks regions and provides decoy contigs for optimal read mapping.
Download your own analysis reference set from the GATK resource bundle. Be certain you are mapping to a version of the genome that hard-masks--replaces with Ns--Y chromosome PARs. Imagine the SHOX of not being able to call variants for pseudoautosomal regions.
3. The challenge alternate contigs presents is a familiar one.
Conceptually概念上 it rewraps包含 and regifts给予 the challenge of calling variants for paralogous旁系同源 regions of the genome. The difference is that alternate contigs encompass包含 sequence that is homologous同源的 as well as highly divergent分岐的 for loci基因座 across a population instead of across a genome. By definition, we cannot easily represent the variants alternate haplotypes generate against the primary assembly. And so GRCh38 arms us with named alternate contigs that beg to be used when we call their variants. How folks家庭 choose to do this with the leeway留有余地 given by VCF specifications will depend on research aims.
4. Latest versions of BWA-MEM handle GRCh38 alternate contig mappings.
You want to map in an alt-aware manner, i.e. you want your alts handled. Without the handling, you’ll just get a bunch of MAPQ zero ghost幽灵 reads mapping to both (i) the primary assembly regions that have alternate contigs and (ii) the homologous alternate contig regions. Just as正像 you cannot eat ghost chips, GATK tools refuse to consider zero (and low) MAPQ alignments. No. You. Do. Not. Want. This. Make sure toupdate to BWA-MEM version 0.7.13+ to be able to map with alt-handling. I’m partial to calling it ghost-busting. This enables two things. First, because it prioritizes( 把…区分优先次序) alignments on the primary assembly by disappearing alignments from the alternate contigs, it effectively lets you avoid redundantly冗余地 calling variants on homologous regions of alternate loci. Second, it allows for an additional postalt-processing step that populates multiple alt loci contig(s) with nonzero MAPQ alignments. This enables super-charged variant calling on all the alt contigs. For details, read BWA’s alt-specific README-alt. Although the README currently is marked for an earlier version of the tool, its concepts still apply.

5. Alt-handling requires the SAM format ALT index file.
Special handling requires a special index file. Alt-handling requires that an ALT index is available with the other BWA indexes. Heng Li provides the ALT index for GRCh38 in the linux bwa.kit v0.7.15. Find the hs38DH.fa.alt file in the resource-GRCh38 folder and explore it using Samtools to confirm the following.
-
3,177 total records
-
792 mapped, of which six are supplementary, that correspond to alternate contigs
- 528 HLA contigs (3 supplementary)
- 264 non-HLA alt contigs (3 supplementary)
Each alternate contig record lists a CIGAR string, some of which are rather convoluted复杂, that aligns the alternate contig back to its primary assembly locus. For six of the alternate contigs, we have two alignments each.
- Leaving us 2,385 unmapped records corresponding to decoy contigs. These exclude the EBV contig, which the index considers a part of the primary assembly.
The decoys contain transposable转座 and alpha satellite卫星 elements including diverged variants. Why are they represented in the ALT index? See the next takeaway.
6. New Tutorial#8017 shows how to map to GRCh38 with alt-handling and then some.
Tutorial#8017 starts with indexing the reference, reiterates重申 the essentiality of the ALT index and then maps in an alt-aware manner using simulated reads to a miniature-reference. It then goes on to show how to postalt-process alignments using the bwa-postalt.js script. The tutorial does not tell you what to do per se, but rather shows what happens when you use certain options. You definitely当然 want to read sections 5–6 if you plan on calling variants on alternate contigs.
During postalt-processing, two reshufflings改组 take place. First, alignments that can map to both a primary locus and an alternate locus are mapped to both with non-zero MAPQ alignments. These multimappers are supplementary on the alt. Second, if an alignment on the primary assembly aligns better on a decoy contig, then its alignment on the primary assembly is deprioritized次优 with a zero MAPQ score. The tutorial gives an example of the first reshuffle. For those interested in seeing the second reshuffle, I have a suggestion. Change the mini-reference’s single ALT index record to mimic模仿 that of a decoy, i.e. change it to an unmapped record, then see what happens when you postalt-process.
If your research aims require one of the reshufflings but not the other, or selective handling for particular loci, then one approach could be to modify the ALT index for the selective postalt-processing.
7. Simulate read mapping for your favorite alternate haplotype.
Tutorial#7859 shows how to generate simulated reads so you can see results akin类似 to those in Tutorial#8017 for your favorite alternate contig. For both tutorials, I use the GPI gene’s singular alternate contig as the example.
Using the liberty自由 the blog博客 format provides, I will digress离题 here. The GPI locus encodes for glucose-6-phosphate isomerase, a protein that has an intracellular role in sugar metabolism and also moonlights extracellularly as Neuroleukin, a factor involved in nerve tissue growth. I chose this locus because (i) it is one of the smallest alternate contigs not near a telomere端粒区, (ii) I used to study metabolism and (iii) I worked on an identically named, unrelated molecule. Yes, really.
So, how significant are the alternate contigs? To start answering this question, I asked another. What story can I find for the GPI locus?
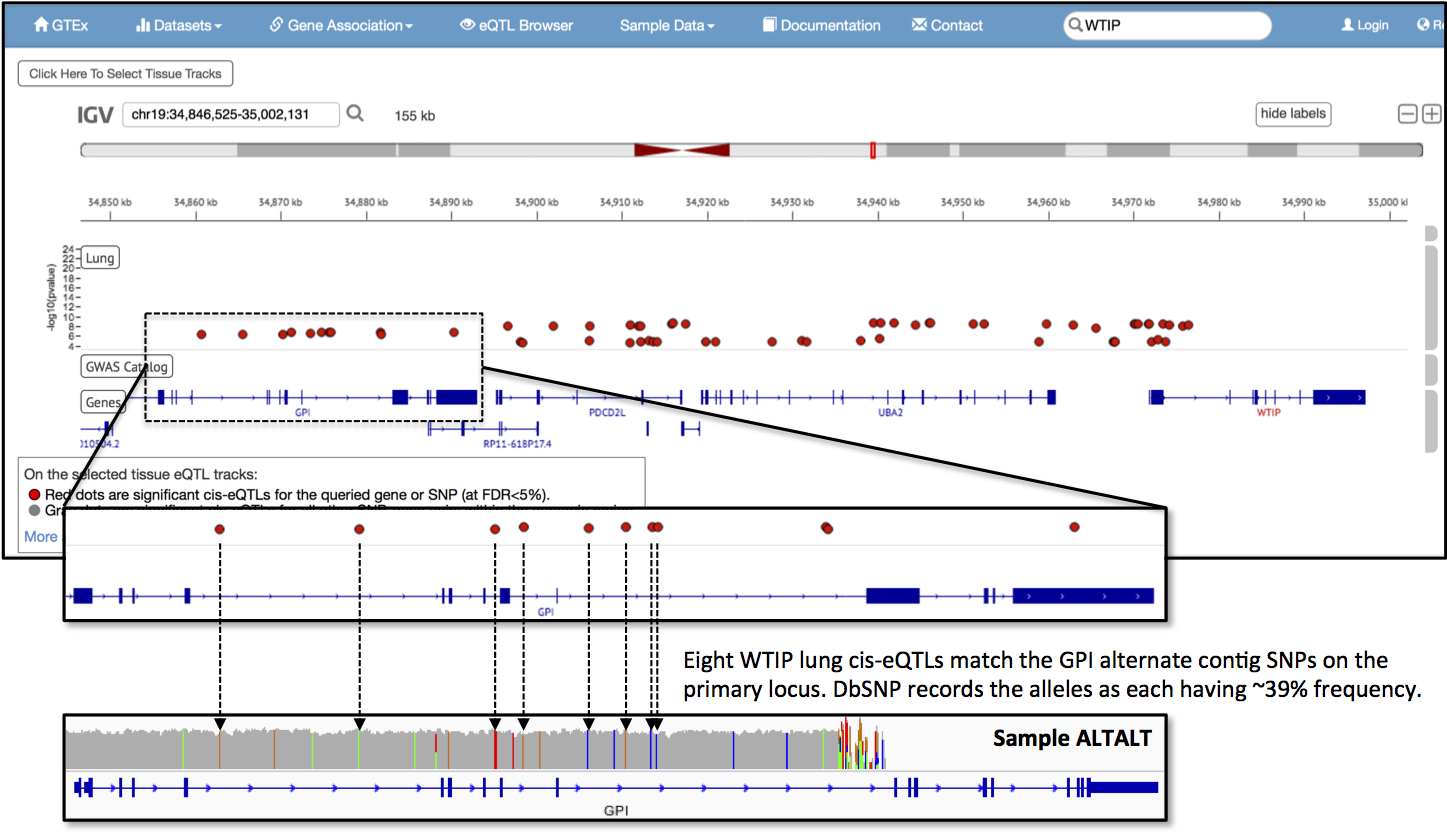
I did a little digging last Saturday afternoon for evidence of the alternate haplotype in data resources. In GTex, a project that measures healthy tissue-specific RNA isoform expression, I found that the GPI locus provides cis-eQTLs for WTIP in lung tissue. WTIP encodes for Wilms tumor 1 interacting protein and is three genes down from the GPI locus. Eight of the 11 eQTL sites on the GPI gene match SNPs that my simulated reads, representing the alternate haplotype, generate on the primary assembly. These sites, when Ilook them up in dbSNP, are all listed as minor alleles and intronic variants. The average global minor allele frequency for the eight SNPs is 38.7% (+/- 0.90%), with 1936 (+/- 45.0) observations in the1000 Genomes Project phase 3 data. It looks like the GPI locus alternate haplotype is not uncommon and it already has some observed associations.
8. Our production workflow for single sample variant calling on GRCh38 is public and uses shiny new features.
Check it out in our Broad pipelines WDL scripts repository. The document describing the workflow has the .md extension in the set named PairedEndSingleSampleWf. Even if you are unfamiliar with what is a WDL, no worries. The document focuses on explaining the data transformation steps from alignment to single-sample SNP and indel variant calling. The workflow maps paired reads in an alt-aware manner to GRCh38 and then uses HaplotypeCaller to generate a GVCF callset for the primary assembly. New features the workflow uses include query-grouped alignments through duplicate marking and addition of NM and UQ tags with SetNmAndUqTags.
9. Finally, there is no better time than now to start learning WDL.
It’s pretty straightforward. Using instructions provided by our WDL documentation, even yours truly has written her first three scripts for Tutorial#8017’s workflows. These we share via our new GATK Tutorials WDL scripts repo. WDL scripts will become more prevalent going forward. In conjunction with Docker, these process-centric pipeline scripts enable better provenance and reproducibility in research. If you are a complete newb to WDL, e.g. don’t know how to pronounce the acronym, then start with Blog#7349.


 浙公网安备 33010602011771号
浙公网安备 33010602011771号