

数据分析与数据挖掘研究之一
前言:之前做过一些数据分析与数据挖掘相关的工作,最近抽空将之前做的内容简单整理一下,方便查看,主要使用R语言和PERL脚本语言,使用TCGA和ICGC数据库中的临床数据,做类似的分析可以参考一下,如果想查看详细内容与数据可以通过本人的Gitee及Github仓库下载,链接于篇尾附上。
一、标题:Effect of HSP90AB1 on the local immune response of hepatocellular carcinoma and it realtionship to prognosis(HSP90β对肝癌局部免疫的影响及对肝癌患者预后的影响)
二、部分代码及结果展示:
1、整理TCGA数据库肝细胞癌临床数据的部分PERL脚本
use strict;
#use warnings;
use XML::Simple;
opendir(RD, ".") or die $!;
my @dirs=readdir(RD);
closedir(RD);
open(WF,">clinical.xls") or die $!;
print WF "Id\tfutime\tfustat\tAge\tGender\tGrade\tStage\tT\tM\tN\n";
foreach my $dir(@dirs){
#print $dir . "\n";
next if($dir eq '.');
next if($dir eq '..');
#print $dir . "\n";
if(-d $dir){
opendir(RD,"$dir") or die $!;
while(my $xmlfile=readdir(RD)){
if($xmlfile=~/\.xml$/){
#print "$dir\\$xmlfile\n";
my $userxs = XML::Simple->new(KeyAttr => "name");
my $userxml="";
if(-f "$dir/$xmlfile"){
$userxml = $userxs->XMLin("$dir/$xmlfile");
}else{
$userxml = $userxs->XMLin("$dir\$xmlfile");
}
# print output
#open(WF,">dumper.txt") or die $!;
#print WF Dumper($userxml);
#close(WF);
my $disease_code=$userxml->{'admin:admin'}{'admin:disease_code'}{'content'}; #get disease code
my $disease_code_lc=lc($disease_code);
my $patient_key=$disease_code_lc . ':patient'; #ucec:patient
my $follow_key=$disease_code_lc . ':follow_ups';
my $patient_barcode=$userxml->{$patient_key}{'shared:bcr_patient_barcode'}{'content'}; #TCGA-AX-A1CJ
my $gender=$userxml->{$patient_key}{'shared:gender'}{'content'}; #male/female
my $age=$userxml->{$patient_key}{'clin_shared:age_at_initial_pathologic_diagnosis'}{'content'};
my $race=$userxml->{$patient_key}{'clin_shared:race_list'}{'clin_shared:race'}{'content'}; #white/black
my $grade=$userxml->{$patient_key}{'shared:neoplasm_histologic_grade'}{'content'}; #G1/G2/G3
my $clinical_stage=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:clinical_stage'}{'content'}; #stage I
my $clinical_T=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:tnm_categories'}{'shared_stage:clinical_categories'}{'shared_stage:clinical_T'}{'content'};
my $clinical_M=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:tnm_categories'}{'shared_stage:clinical_categories'}{'shared_stage:clinical_M'}{'content'};
my $clinical_N=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:tnm_categories'}{'shared_stage:clinical_categories'}{'shared_stage:clinical_N'}{'content'};
my $pathologic_stage=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:pathologic_stage'}{'content'}; #stage I
my $pathologic_T=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:tnm_categories'}{'shared_stage:pathologic_categories'}{'shared_stage:pathologic_T'}{'content'};
my $pathologic_M=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:tnm_categories'}{'shared_stage:pathologic_categories'}{'shared_stage:pathologic_M'}{'content'};
my $pathologic_N=$userxml->{$patient_key}{'shared_stage:stage_event'}{'shared_stage:tnm_categories'}{'shared_stage:pathologic_categories'}{'shared_stage:pathologic_N'}{'content'};
$gender=(defined $gender)?$gender:"unknow";
$age=(defined $age)?$age:"unknow";
$race=(defined $race)?$race:"unknow";
$grade=(defined $grade)?$grade:"unknow";
$clinical_stage=(defined $clinical_stage)?$clinical_stage:"unknow";
$clinical_T=(defined $clinical_T)?$clinical_T:"unknow";
$clinical_M=(defined $clinical_M)?$clinical_M:"unknow";
$clinical_N=(defined $clinical_N)?$clinical_N:"unknow";
$pathologic_stage=(defined $pathologic_stage)?$pathologic_stage:"unknow";
$pathologic_T=(defined $pathologic_T)?$pathologic_T:"unknow";
$pathologic_M=(defined $pathologic_M)?$pathologic_M:"unknow";
$pathologic_N=(defined $pathologic_N)?$pathologic_N:"unknow";
my $survivalTime="";
my $vital_status=$userxml->{$patient_key}{'clin_shared:vital_status'}{'content'};
my $followup=$userxml->{$patient_key}{'clin_shared:days_to_last_followup'}{'content'};
my $death=$userxml->{$patient_key}{'clin_shared:days_to_death'}{'content'};
if($vital_status eq 'Alive'){
$survivalTime="$followup\t0";
}
else{
$survivalTime="$death\t1";
}
for my $i(keys %{$userxml->{$patient_key}{$follow_key}}){
eval{
$followup=$userxml->{$patient_key}{$follow_key}{$i}{'clin_shared:days_to_last_followup'}{'content'};
$vital_status=$userxml->{$patient_key}{$follow_key}{$i}{'clin_shared:vital_status'}{'content'};
$death=$userxml->{$patient_key}{$follow_key}{$i}{'clin_shared:days_to_death'}{'content'};
};
if($@){
for my $j(0..5){ #假设最多有6次随访
my $followup_for=$userxml->{$patient_key}{$follow_key}{$i}[$j]{'clin_shared:days_to_last_followup'}{'content'};
my $vital_status_for=$userxml->{$patient_key}{$follow_key}{$i}[$j]{'clin_shared:vital_status'}{'content'};
my $death_for=$userxml->{$patient_key}{$follow_key}{$i}[$j]{'clin_shared:days_to_death'}{'content'};
if( ($followup_for =~ /\d+/) || ($death_for =~ /\d+/) ){
$followup=$followup_for;
$vital_status=$vital_status_for;
$death=$death_for;
my @survivalArr=split(/\t/,$survivalTime);
if($vital_status eq 'Alive'){
if($followup>$survivalArr[0]){
$survivalTime="$followup\t0";
}
}
else{
if($death>$survivalArr[0]){
$survivalTime="$death\t1";
}
}
}
}
}
my @survivalArr=split(/\t/,$survivalTime);
if($vital_status eq 'Alive'){
if($followup>$survivalArr[0]){
$survivalTime="$followup\t0";
}
}
else{
if($death>$survivalArr[0]){
$survivalTime="$death\t1";
}
}
}
print WF "$patient_barcode\t$survivalTime\t$age\t$gender\t$grade\t$pathologic_stage\t$pathologic_T\t$pathologic_M\t$pathologic_N\n";
}
}
close(RD);
}
}
close(WF);
2、使用R语言分析正常组与肿瘤组中HSP90AB1的表达情况
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("limma")
#install.packages("ggplot2")
#install.packages("ggpubr")
#引用包
library(limma)
library(ggplot2)
library(ggpubr)
expFile="symbol.txt" #表达输入文件
gene="VCAN" #基因的名称
setwd("C:\\Users\\lexb4\\Desktop\\geneImmune\\07.diff") #设置工作目录
#读取基因表达文件,并对数据进行处理
rt=read.table(expFile, header=T, sep="\t", check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)), nrow=nrow(exp), dimnames=dimnames)
data=avereps(data)
data=t(data[gene,,drop=F])
#正常和肿瘤数目
group=sapply(strsplit(rownames(data),"\\-"), "[", 4)
group=sapply(strsplit(group,""), "[", 1)
group=gsub("2", "1", group)
conNum=length(group[group==1]) #正常组样品数目
treatNum=length(group[group==0]) #肿瘤组样品数目
Type=c(rep(1,conNum), rep(2,treatNum))
#差异分析
exp=cbind(data, Type)
exp=as.data.frame(exp)
colnames(exp)=c("gene", "Type")
exp$Type=ifelse(exp$Type==1, "Normal", "Tumor")
exp$gene=log2(exp$gene+1)
#设置比较组
group=levels(factor(exp$Type))
exp$Type=factor(exp$Type, levels=group)
comp=combn(group,2)
my_comparisons=list()
for(i in 1:ncol(comp)){my_comparisons[[i]]<-comp[,i]}
#绘制boxplot
boxplot=ggboxplot(exp, x="Type", y="gene", color="Type",
xlab="",
ylab=paste0(gene, " expression"),
legend.title="Type",
palette = c("blue","red"),
add = "jitter")+
stat_compare_means(comparisons=my_comparisons,symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1), symbols = c("***", "**", "*", "ns")),label = "p.signif")
#输出图片
pdf(file=paste0(gene,".diff.pdf"), width=5, height=4.5)
print(boxplot)
dev.off()
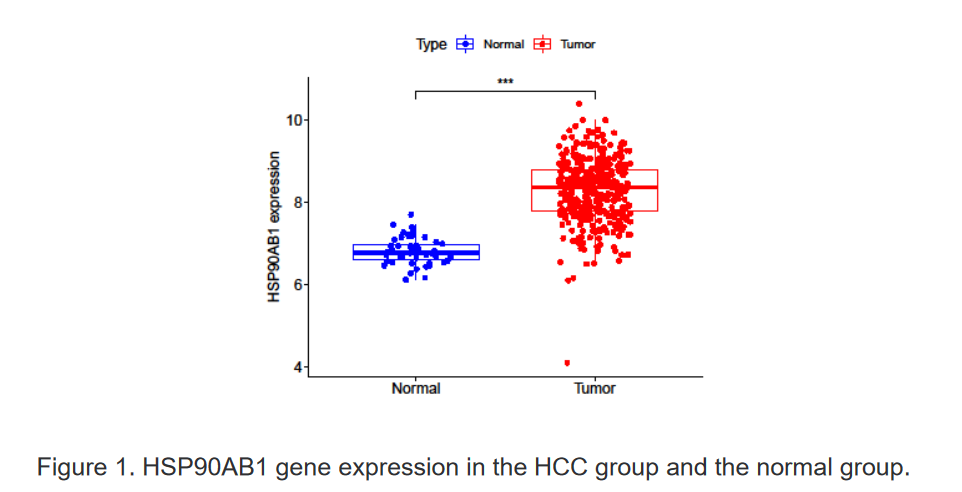
3、使用R语言分析不同类型免疫细胞在肝细胞癌中的表达水平及相关关系
#install.packages("corrplot")
library(corrplot) #引用包
immFile="CIBERSORT-Results.txt" #免疫细胞浸润的结果文件
pFilter=0.05 #免疫细胞浸润结果过滤条件
setwd("C:\\Users\\Administrator\\Desktop\\geneimmune\\10immunePlot") #设置工作目录
#读取免疫细胞浸润的结果文件,并对数据进行整理
immune=read.table(immFile, header=T, sep="\t", check.names=F, row.names=1)
immune=immune[immune[,"P-value"]<pFilter,]
immune=as.matrix(immune[,1:(ncol(immune)-3)])
data=t(immune)
#绘制柱状图
col=rainbow(nrow(data), s=0.7, v=0.7)
pdf(file="barplot.pdf", width=22, height=10)
par(las=1,mar=c(8,5,4,16),mgp=c(3,0.1,0),cex.axis=1.5)
a1=barplot(data,col=col,yaxt="n",ylab="Relative Percent",xaxt="n",cex.lab=1.8)
a2=axis(2,tick=F,labels=F)
axis(2,a2,paste0(a2*100,"%"))
axis(1,a1,labels=F)
par(srt=60,xpd=T);text(a1,-0.02,colnames(data),adj=1,cex=0.6);par(srt=0)
ytick2=cumsum(data[,ncol(data)]);ytick1=c(0,ytick2[-length(ytick2)])
legend(par('usr')[2]*0.98,par('usr')[4],legend=rownames(data),col=col,pch=15,bty="n",cex=1.3)
dev.off()
#删除正常样品
group=sapply(strsplit(colnames(data),"\\-"), "[", 4)
group=sapply(strsplit(group,""), "[", 1)
group=gsub("2", "1", group)
data=data[,group==0,drop=F]
#绘制免疫细胞相关性的图形
pdf(file="corrplot.pdf", width=13, height=13)
par(oma=c(0.5,1,1,1.2))
immune=immune[,colMeans(immune)>0]
M=cor(immune)
corrplot(M,
method = "color",
order = "hclust",
tl.col="black",
addCoef.col = "black",
number.cex = 0.8,
col=colorRampPalette(c("blue", "white", "red"))(50)
)
dev.off()

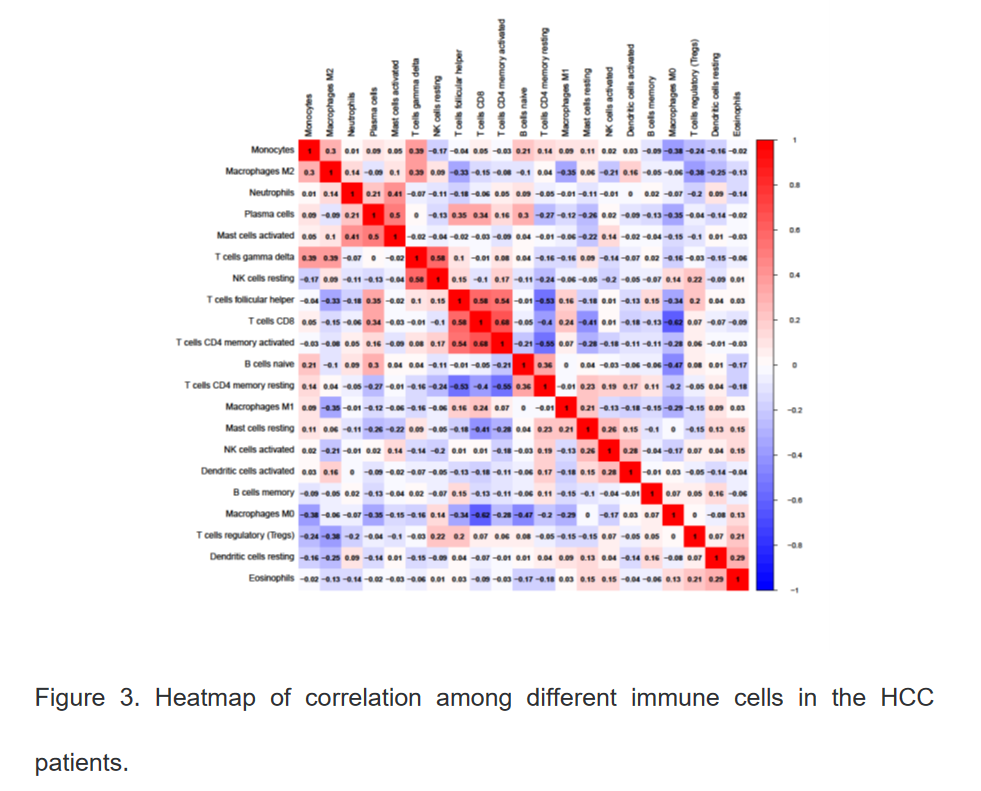
4、使用R语言分析正常组及肝癌组中不同免疫细胞浸润水平
#install.packages("pheatmap")
#install.packages("vioplot")
#引用包
library(vioplot)
library(pheatmap)
input="CIBERSORT-Results.txt" #免疫细胞浸润文件
pFilter=0.05 #免疫细胞浸润结果过滤条件
setwd("C:\\Users\\Administrator\\Desktop\\生信文章\\geneimmune\\11heatmap\\vioplot-high") #设置工作目录
#读取免疫结果文件,并对数据进行整理
immune=read.table("CIBERSORT-Results.txt", header=T, sep="\t", check.names=F, row.names=1)
immune=immune[immune[,"P-value"]<pFilter,,drop=F]
immune=as.matrix(immune[,1:(ncol(immune)-3)])
data=t(immune)
#正常和肿瘤数目
group=sapply(strsplit(colnames(data),"\\-"), "[", 4)
group=sapply(strsplit(group,""), "[", 1)
group=gsub("2", "1", group)
conNum=length(group[group==1]) #正常组样品数目
treatNum=length(group[group==0]) #肿瘤组样品数目
#定义热图的注释文件
Type=c(rep("Normal",conNum), rep("Tumor",treatNum))
names(Type)=colnames(data)
Type=as.data.frame(Type)
#绘制热图
pdf(file="heatmap.pdf", width=12, height=6)
pheatmap(data,
annotation=Type,
color = colorRampPalette(c(rep("green",1), rep("black",1), rep("red",3)))(100),
cluster_cols =F,
show_colnames=F,
fontsize = 8,
fontsize_row=7,
fontsize_col=5)
dev.off()
#绘制小提琴图
data=t(data)
outTab=data.frame()
pdf(file="vioplot.pdf", width=13, height=8)
par(las=1, mar=c(10,6,3,3))
x=c(1:ncol(data))
y=c(1:ncol(data))
xMax=ncol(data)*3-2
plot(x,y,
xlim=c(0,xMax),ylim=c(min(data),max(data)+0.02),
main="", xlab="", ylab="Fraction",
pch=21,
col="white",
xaxt="n")
#对每个免疫细胞循环,绘制小提琴图,正常样品用绿色表示,肿瘤样品用红色表示
for(i in 1:ncol(data)){
if(sd(data[1:conNum,i])==0){
data[1,i]=0.00001
}
if(sd(data[(conNum+1):(conNum+treatNum),i])==0){
data[(conNum+1),i]=0.00001
}
conData=data[1:conNum,i]
treatData=data[(conNum+1):(conNum+treatNum),i]
vioplot(conData,at=3*(i-1),lty=1,add = T,col = 'green')
vioplot(treatData,at=3*(i-1)+1,lty=1,add = T,col = 'red')
wilcoxTest=wilcox.test(conData, treatData)
p=wilcoxTest$p.value
if(p<pFilter){
cellPvalue=cbind(Cell=colnames(data)[i], pvalue=p)
outTab=rbind(outTab, cellPvalue)
}
mx=max(c(conData,treatData))
lines(c(x=3*(i-1)+0.2,x=3*(i-1)+0.8),c(mx,mx))
text(x=3*(i-1)+0.5, y=mx+0.02, labels=ifelse(p<0.001, paste0("p<0.001"), paste0("p=",sprintf("%.03f",p))), cex = 0.8)
}
legend("topright",
c("Normal", "Tumor"),
lwd=5,bty="n",cex=1.2,
col=c("green","red"))
text(seq(1,xMax,3),-0.05,xpd = NA,labels=colnames(data),cex = 1,srt = 45,pos=2)
dev.off()
#输出免疫细胞和p值表格文件
write.table(outTab,file="diff.result.txt",sep="\t",row.names=F,quote=F)

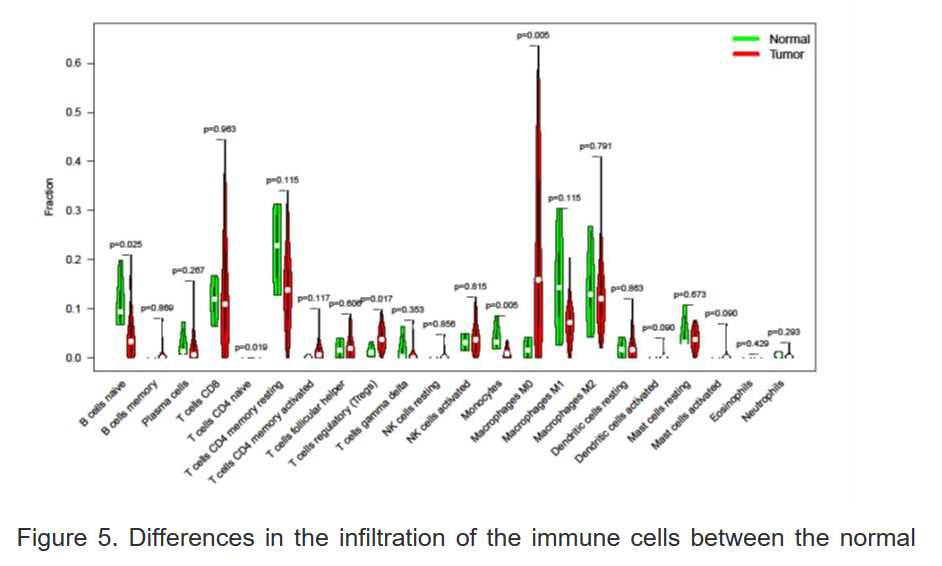
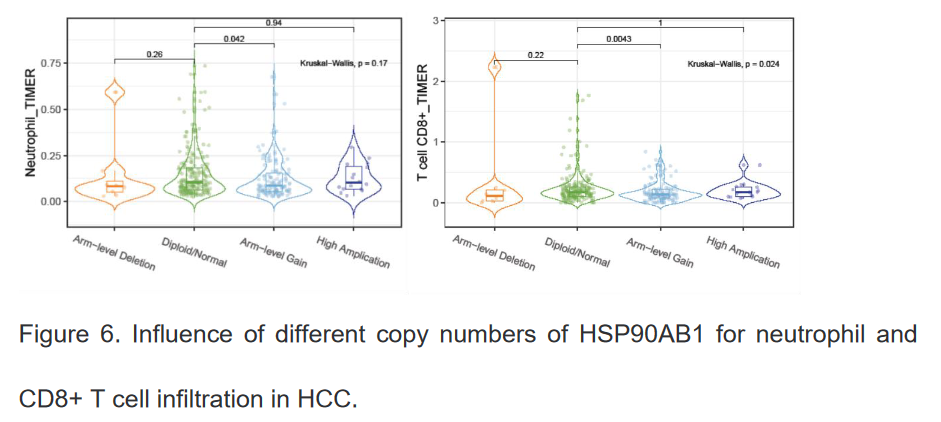
5、不同拷贝子数目的HSP90β对中性粒细胞和CD8阳性T细胞在肝癌局部浸润的影响
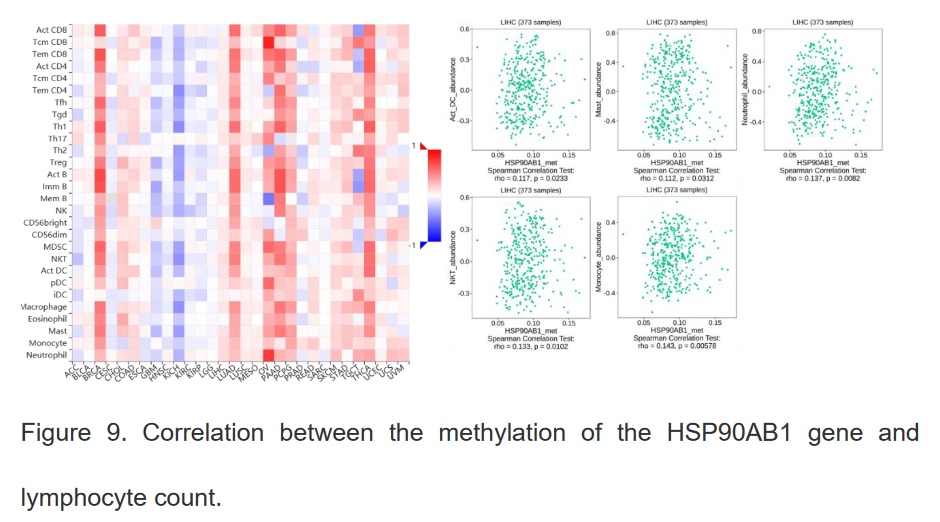
6、HSP90β基因的表达水平、拷贝子水平及甲基化水平与不同淋巴细胞数量之间的关系


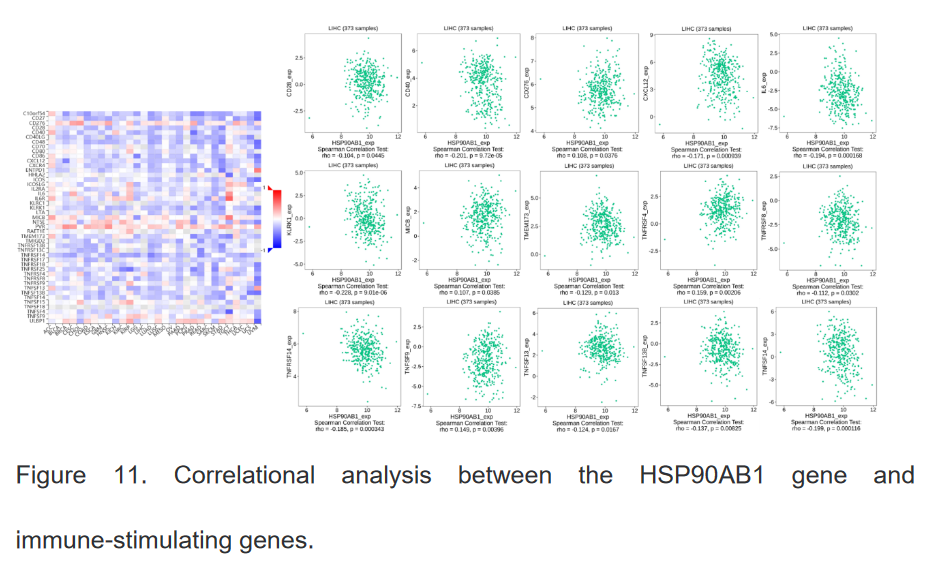
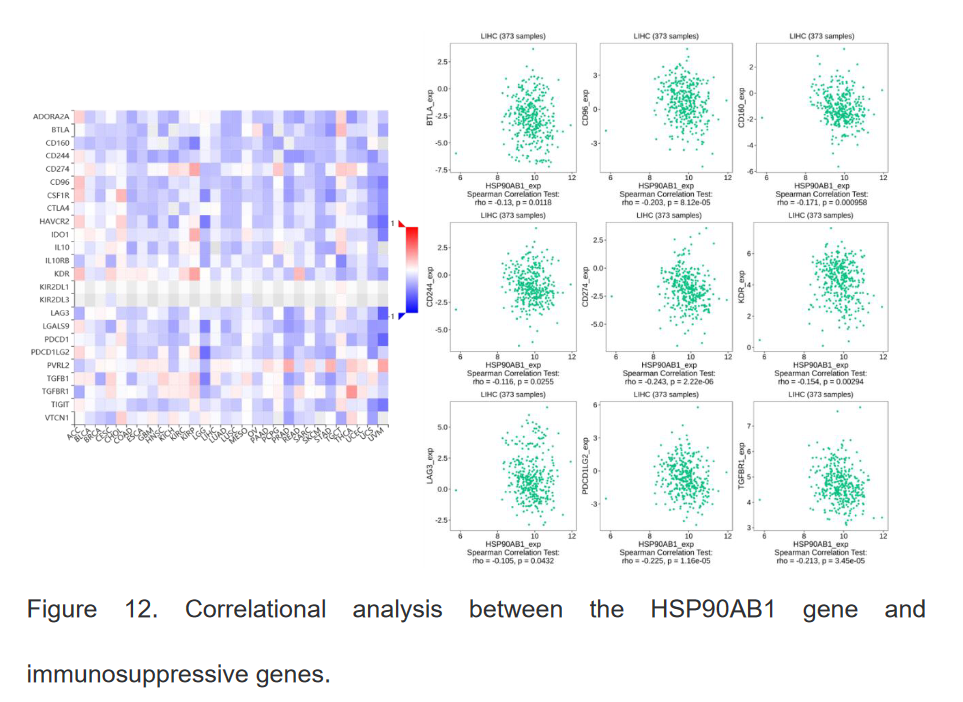
7、HSP90β基因与免疫调节基因之间的关系
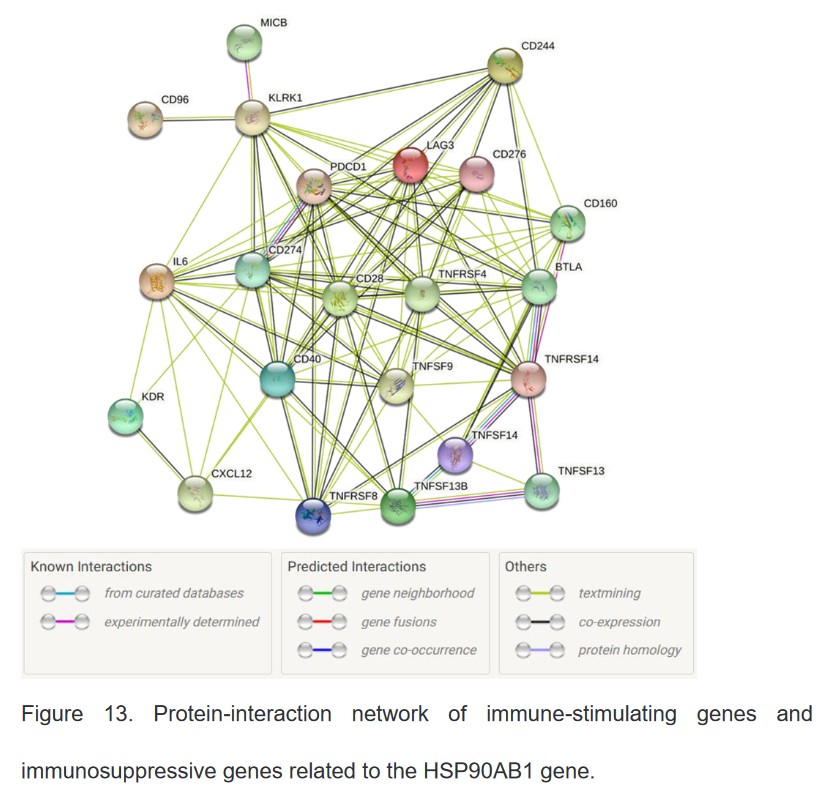
8、与HSP90β相关的免疫调节基因的蛋白互作网络
9、GO及相关通路分析
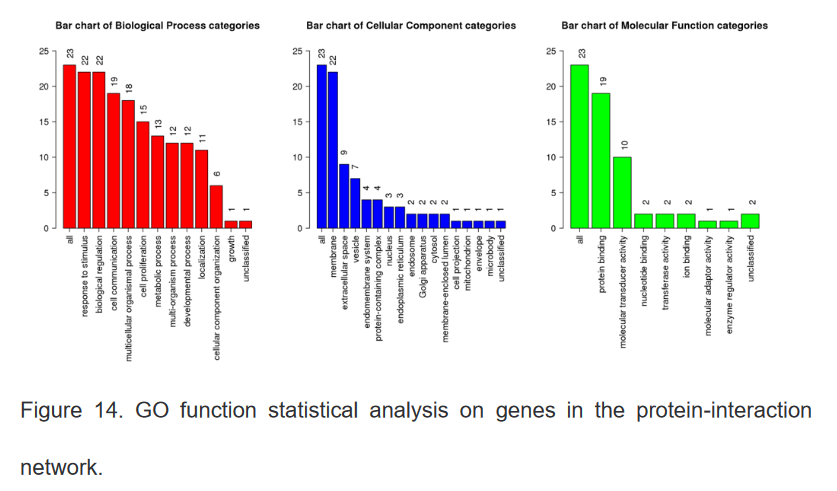

10、整理患者基因表达水平与临床生存信息
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("limma")
library(limma) #引用包
expFile="symbol.txt" #表达数据文件
cliFile="time.txt" #临床数据文件
geneFile="gene.txt" #基因列表文件
setwd("C:\\Users\\Administrator\\Desktop\\geneimmune\\24mergeTime") #工作目录(需修改)
#读取表达文件,并对输入文件整理
rt=read.table(expFile, header=T, sep="\t", check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp), colnames(exp))
data=matrix(as.numeric(as.matrix(exp)), nrow=nrow(exp), dimnames=dimnames)
data=avereps(data)
data=data[rowMeans(data)>0,]
#读取免疫基因的表达量
gene=read.table(geneFile, header=F, sep="\t", check.names=F)
sameGene=intersect(as.vector(gene[,1]), row.names(data))
data=data[sameGene,]
#删掉正常样品
group=sapply(strsplit(colnames(data),"\\-"), "[", 4)
group=sapply(strsplit(group,""), "[", 1)
group=gsub("2", "1", group)
data=data[,group==0]
colnames(data)=gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", colnames(data))
data=t(data)
data=avereps(data)
#读取生存数据
cli=read.table(cliFile, header=T, sep="\t", check.names=F, row.names=1) #读取临床文件
#数据合并并输出结果
sameSample=intersect(row.names(data), row.names(cli))
data=data[sameSample,]
cli=cli[sameSample,]
out=cbind(cli,data)
out=cbind(id=row.names(out),out)
write.table(out,file="expTime.txt",sep="\t",row.names=F,quote=F)
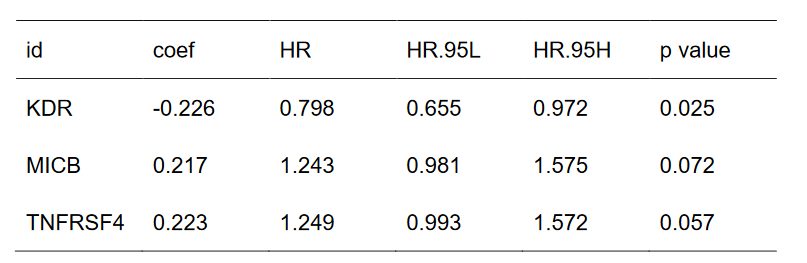
11、筛选HSP90β蛋白互作网络中预后相关的免疫调节基因,绘制森林图
#install.packages('survival')
library(survival) #引用包
coxPfilter=0.05 #显著性过滤标准
inputFile="expTime.txt" #输入文件
setwd("C:\\Users\\Administrator\\Desktop\\geneimmune\\25uniCox") #设置工作目录
rt=read.table(inputFile, header=T, sep="\t", check.names=F, row.names=1) #读取输入文件
rt$futime=rt$futime/365
rt[,3:ncol(rt)]=log2(rt[,3:ncol(rt)]+1)
#对基因进行循环,找出预后相关的基因
outTab=data.frame()
sigGenes=c("futime","fustat")
for(i in colnames(rt[,3:ncol(rt)])){
#cox分析
cox <- coxph(Surv(futime, fustat) ~ rt[,i], data = rt)
coxSummary = summary(cox)
coxP=coxSummary$coefficients[,"Pr(>|z|)"]
#保留预后相关的基因
if(coxP<coxPfilter){
sigGenes=c(sigGenes,i)
outTab=rbind(outTab,
cbind(id=i,
HR=coxSummary$conf.int[,"exp(coef)"],
HR.95L=coxSummary$conf.int[,"lower .95"],
HR.95H=coxSummary$conf.int[,"upper .95"],
pvalue=coxSummary$coefficients[,"Pr(>|z|)"])
)
}
}
#输出单因素的结果
write.table(outTab,file="uniCox.txt",sep="\t",row.names=F,quote=F)
#输出单因素显著基因的表达量
uniSigExp=rt[,sigGenes]
uniSigExp=cbind(id=row.names(uniSigExp),uniSigExp)
write.table(uniSigExp,file="uniSigExp.txt",sep="\t",row.names=F,quote=F)
############绘制森林图函数############
bioForest=function(coxFile=null, forestFile=null, forestCol=null){
#读取输入文件
rt <- read.table(coxFile, header=T, sep="\t", check.names=F, row.names=1)
gene <- rownames(rt)
hr <- sprintf("%.3f",rt$"HR")
hrLow <- sprintf("%.3f",rt$"HR.95L")
hrHigh <- sprintf("%.3f",rt$"HR.95H")
Hazard.ratio <- paste0(hr,"(",hrLow,"-",hrHigh,")")
pVal <- ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue))
#输出图形
pdf(file=forestFile, width=6.5, height=5)
n <- nrow(rt)
nRow <- n+1
ylim <- c(1,nRow)
layout(matrix(c(1,2),nc=2),width=c(3,2.5))
#绘制森林图左边的基因信息
xlim = c(0,3)
par(mar=c(4,2.5,2,1))
plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,xlab="",ylab="")
text.cex=0.8
text(0,n:1,gene,adj=0,cex=text.cex)
text(1.5-0.5*0.2,n:1,pVal,adj=1,cex=text.cex);text(1.5-0.5*0.2,n+1,'pvalue',cex=text.cex,font=2,adj=1)
text(3,n:1,Hazard.ratio,adj=1,cex=text.cex);text(3,n+1,'Hazard ratio',cex=text.cex,font=2,adj=1,)
#绘制森林图
par(mar=c(4,1,2,1),mgp=c(2,0.5,0))
xlim = c(0,max(as.numeric(hrLow),as.numeric(hrHigh)))
plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,ylab="",xaxs="i",xlab="Hazard ratio")
arrows(as.numeric(hrLow),n:1,as.numeric(hrHigh),n:1,angle=90,code=3,length=0.05,col="darkblue",lwd=2.5)
abline(v=1,col="black",lty=2,lwd=2)
boxcolor = ifelse(as.numeric(hr) > 1, forestCol[1], forestCol[2])
points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex=1.6)
axis(1)
dev.off()
}
bioForest(coxFile="uniCox.txt", forestFile="forest.pdf", forestCol=c("red","green"))
12、使用筛选出的基因构建预后模型
#install.packages("glmnet")
#install.packages("survival")
#install.packages('survminer')
#引用包
library(glmnet)
library(survival)
library(survminer)
inputFile="uniSigExp.txt" #单因素显著基因的表达输入文件
setwd("C:\\Users\\lexb4\\Desktop\\geneImmune\\26.model") #设置工作目录
rt=read.table(inputFile, header=T, sep="\t", row.names=1, check.names=F) #读取输入文件
#COX模型构建
multiCox=coxph(Surv(futime, fustat) ~ ., data = rt)
multiCox=step(multiCox, direction="both")
multiCoxSum=summary(multiCox)
#输出模型相关信息
outMultiTab=data.frame()
outMultiTab=cbind(
coef=multiCoxSum$coefficients[,"coef"],
HR=multiCoxSum$conf.int[,"exp(coef)"],
HR.95L=multiCoxSum$conf.int[,"lower .95"],
HR.95H=multiCoxSum$conf.int[,"upper .95"],
pvalue=multiCoxSum$coefficients[,"Pr(>|z|)"])
outMultiTab=cbind(id=row.names(outMultiTab),outMultiTab)
write.table(outMultiTab, file="multiCox.txt", sep="\t", row.names=F, quote=F)
#输出风险文件
score=predict(multiCox, type="risk", newdata=rt)
coxGene=rownames(multiCoxSum$coefficients)
coxGene=gsub("`", "", coxGene)
outCol=c("futime", "fustat", coxGene)
risk=as.vector(ifelse(score>median(score), "high", "low"))
outTab=cbind(rt[,outCol], riskScore=as.vector(score), risk)
write.table(cbind(id=rownames(outTab),outTab), file="risk.txt", sep="\t", quote=F, row.names=F)
#绘制森林图
pdf(file="multi.forest.pdf", width=10, height=6, onefile=FALSE)
ggforest(multiCox,
data=rt,
main = "Hazard ratio",
cpositions = c(0.02,0.22, 0.4),
fontsize = 0.7,
refLabel = "reference",
noDigits = 2)
dev.off()
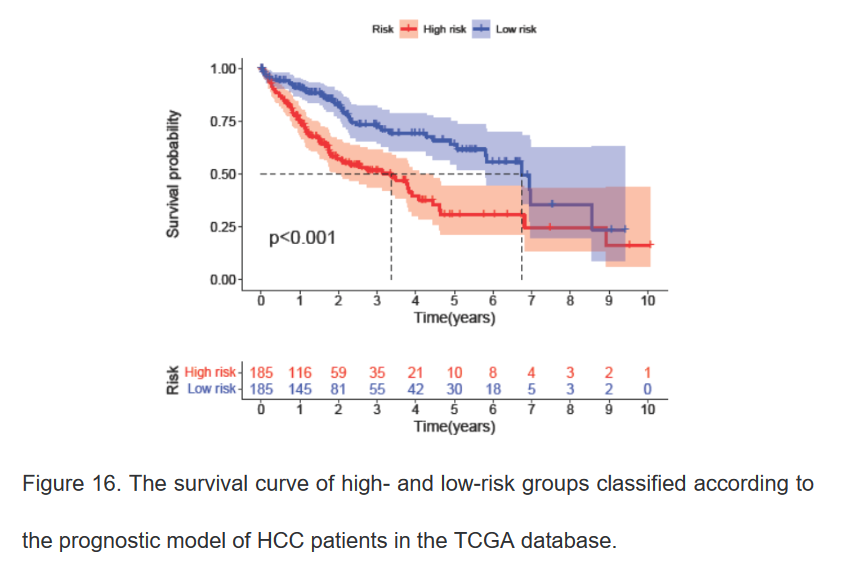
13、绘制该预后模型高低风险组的生存曲线
#install.packages("survival")
#install.packages("survminer")
#引用包
library(survival)
library(survminer)
setwd("C:\\Users\\lexb4\\Desktop\\geneImmune\\27.survival") #设置工作目录
#定义生存曲线的函数
bioSurvival=function(inputFile=null, outFile=null){
#读取输入文件
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
#比较高低风险组生存差异,得到显著性p值
diff=survdiff(Surv(futime, fustat) ~ risk, data=rt)
pValue=1-pchisq(diff$chisq, df=1)
if(pValue<0.001){
pValue="p<0.001"
}else{
pValue=paste0("p=",sprintf("%.03f",pValue))
}
fit <- survfit(Surv(futime, fustat) ~ risk, data = rt)
#print(surv_median(fit))
#绘制生存曲线
surPlot=ggsurvplot(fit,
data=rt,
conf.int=T,
pval=pValue,
pval.size=6,
surv.median.line = "hv",
legend.title="Risk",
legend.labs=c("High risk", "Low risk"),
xlab="Time(years)",
break.time.by = 1,
palette=c("red", "blue"),
risk.table=TRUE,
risk.table.title="",
risk.table.col = "strata",
risk.table.height=.25)
pdf(file=outFile, onefile=FALSE, width=6.5, height=5.5)
print(surPlot)
dev.off()
}
#调用函数,绘制生存曲线
bioSurvival(inputFile="risk.txt", outFile="survival.pdf")
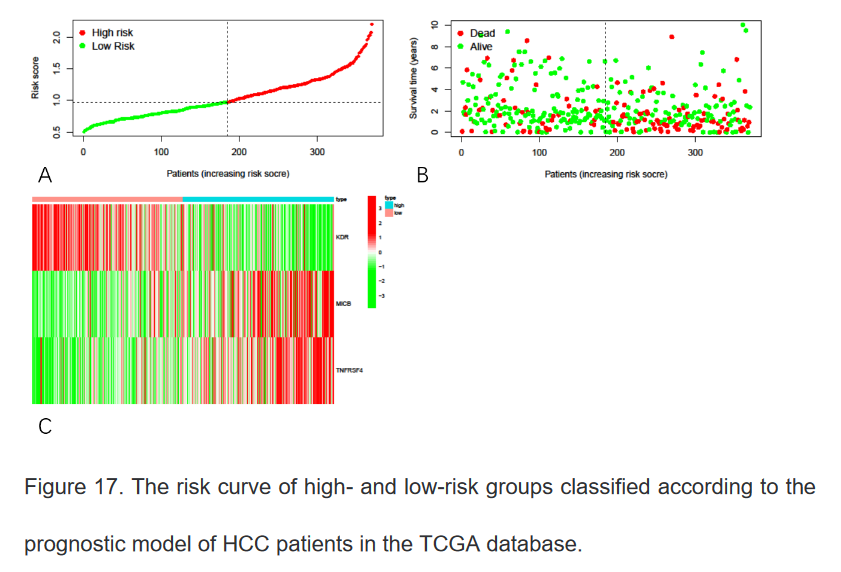
14、绘制不同的风险曲线
#install.packages("pheatmap")
library(pheatmap) #引用包
setwd("C:\\Users\\Administrator\\Desktop\\geneimmune\\28riskPlot") #设置工作目录
#定义风险曲线的函数
bioRiskPlot=function(inputFile=null, riskScoreFile=null, survStatFile=null, heatmapFile=null){
rt=read.table(inputFile, header=T, sep="\t", check.names=F, row.names=1) #读取输入文件
rt=rt[order(rt$riskScore),] #按照风险打分对样品排序
#绘制风险曲线
riskClass=rt[,"risk"]
lowLength=length(riskClass[riskClass=="low"])
highLength=length(riskClass[riskClass=="high"])
lowMax=max(rt$riskScore[riskClass=="low"])
line=rt[,"riskScore"]
line[line>10]=10
pdf(file=riskScoreFile, width=7, height=4)
plot(line, type="p", pch=20,
xlab="Patients (increasing risk socre)", ylab="Risk score",
col=c(rep("green",lowLength),rep("red",highLength)) )
abline(h=lowMax,v=lowLength,lty=2)
legend("topleft", c("High risk", "Low Risk"),bty="n",pch=19,col=c("red","green"),cex=1.2)
dev.off()
#绘制生存状态图
color=as.vector(rt$fustat)
color[color==1]="red"
color[color==0]="green"
pdf(file=survStatFile, width=7, height=4)
plot(rt$futime, pch=19,
xlab="Patients (increasing risk socre)", ylab="Survival time (years)",
col=color)
legend("topleft", c("Dead", "Alive"),bty="n",pch=19,col=c("red","green"),cex=1.2)
abline(v=lowLength,lty=2)
dev.off()
#绘制风险热图
rt1=rt[c(3:(ncol(rt)-2))]
rt1=t(rt1)
annotation=data.frame(type=rt[,ncol(rt)])
rownames(annotation)=rownames(rt)
pdf(file=heatmapFile, width=7, height=4)
pheatmap(rt1,
annotation=annotation,
cluster_cols = FALSE,
cluster_rows = FALSE,
show_colnames = F,
scale="row",
color = colorRampPalette(c(rep("green",3), "white", rep("red",3)))(50),
fontsize_col=3,
fontsize=7,
fontsize_row=8)
dev.off()
}
#调用函数,绘制风险曲线
bioRiskPlot(inputFile="risk.txt",
riskScoreFile="riskScore.pdf",
survStatFile="survStat.pdf",
heatmapFile="heatmap.pdf")
15、绘制不同风险因素森林图比较,并进行预后分析
#install.packages('survival')
library(survival) #引用包
setwd("C:\\Users\\Administrator\\Desktop\\geneimmune\\29indep") #设置工作目录
############绘制森林图函数############
bioForest=function(coxFile=null, forestFile=null, forestCol=null){
#读取输入文件
rt <- read.table(coxFile, header=T, sep="\t", check.names=F, row.names=1)
gene <- rownames(rt)
hr <- sprintf("%.3f",rt$"HR")
hrLow <- sprintf("%.3f",rt$"HR.95L")
hrHigh <- sprintf("%.3f",rt$"HR.95H")
Hazard.ratio <- paste0(hr,"(",hrLow,"-",hrHigh,")")
pVal <- ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue))
#输出图形
pdf(file=forestFile, width=6.5, height=4.5)
n <- nrow(rt)
nRow <- n+1
ylim <- c(1,nRow)
layout(matrix(c(1,2),nc=2),width=c(3,2.5))
#绘制森林图左边的临床信息
xlim = c(0,3)
par(mar=c(4,2.5,2,1))
plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,xlab="",ylab="")
text.cex=0.8
text(0,n:1,gene,adj=0,cex=text.cex)
text(1.5-0.5*0.2,n:1,pVal,adj=1,cex=text.cex);text(1.5-0.5*0.2,n+1,'pvalue',cex=text.cex,font=2,adj=1)
text(3.1,n:1,Hazard.ratio,adj=1,cex=text.cex);text(3.1,n+1,'Hazard ratio',cex=text.cex,font=2,adj=1)
#绘制右边的森林图
par(mar=c(4,1,2,1),mgp=c(2,0.5,0))
xlim = c(0,max(as.numeric(hrLow),as.numeric(hrHigh)))
plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,ylab="",xaxs="i",xlab="Hazard ratio")
arrows(as.numeric(hrLow),n:1,as.numeric(hrHigh),n:1,angle=90,code=3,length=0.05,col="darkblue",lwd=3)
abline(v=1, col="black", lty=2, lwd=2)
boxcolor = ifelse(as.numeric(hr) > 1, forestCol, forestCol)
points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex=2)
axis(1)
dev.off()
}
############绘制森林图函数############
#定义独立预后分析函数
indep=function(riskFile=null,cliFile=null,uniOutFile=null,multiOutFile=null,uniForest=null,multiForest=null){
risk=read.table(riskFile, header=T, sep="\t", check.names=F, row.names=1) #读取风险文件
cli=read.table(cliFile, header=T, sep="\t", check.names=F, row.names=1) #读取临床文件
#数据合并
sameSample=intersect(row.names(cli),row.names(risk))
risk=risk[sameSample,]
cli=cli[sameSample,]
rt=cbind(futime=risk[,1], fustat=risk[,2], cli, riskScore=risk[,(ncol(risk)-1)])
#单因素独立预后分析
uniTab=data.frame()
for(i in colnames(rt[,3:ncol(rt)])){
cox <- coxph(Surv(futime, fustat) ~ rt[,i], data = rt)
coxSummary = summary(cox)
uniTab=rbind(uniTab,
cbind(id=i,
HR=coxSummary$conf.int[,"exp(coef)"],
HR.95L=coxSummary$conf.int[,"lower .95"],
HR.95H=coxSummary$conf.int[,"upper .95"],
pvalue=coxSummary$coefficients[,"Pr(>|z|)"])
)
}
write.table(uniTab,file=uniOutFile,sep="\t",row.names=F,quote=F)
bioForest(coxFile=uniOutFile, forestFile=uniForest, forestCol="green")
#多因素独立预后分析
uniTab=uniTab[as.numeric(uniTab[,"pvalue"])<1,]
rt1=rt[,c("futime", "fustat", as.vector(uniTab[,"id"]))]
multiCox=coxph(Surv(futime, fustat) ~ ., data = rt1)
multiCoxSum=summary(multiCox)
multiTab=data.frame()
multiTab=cbind(
HR=multiCoxSum$conf.int[,"exp(coef)"],
HR.95L=multiCoxSum$conf.int[,"lower .95"],
HR.95H=multiCoxSum$conf.int[,"upper .95"],
pvalue=multiCoxSum$coefficients[,"Pr(>|z|)"])
multiTab=cbind(id=row.names(multiTab),multiTab)
write.table(multiTab,file=multiOutFile,sep="\t",row.names=F,quote=F)
bioForest(coxFile=multiOutFile, forestFile=multiForest, forestCol="red")
}
#独立预后分析
indep(riskFile="risk.txt",
cliFile="clinical.txt",
uniOutFile="uniCox.txt",
multiOutFile="multiCox.txt",
uniForest="uniForest.pdf",
multiForest="multiForest.pdf")
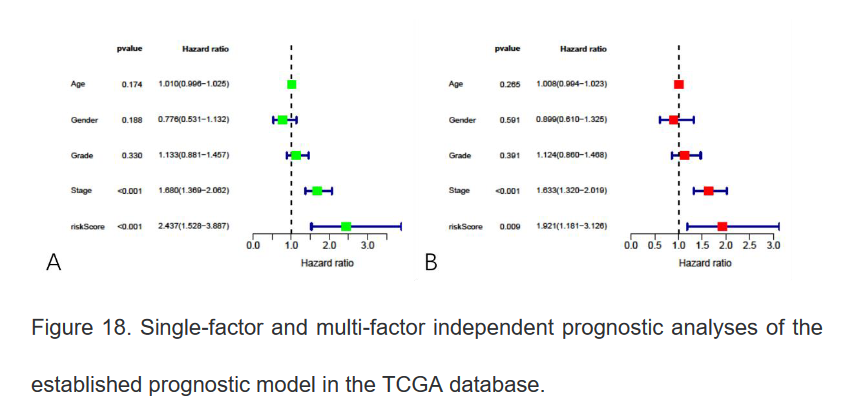
16、绘制ROC曲线
#install.packages("survival")
#install.packages("survminer")
#install.packages("timeROC")
#引用包
library(survival)
library(survminer)
library(timeROC)
riskFile="risk.txt" #风险输入文件
cliFile="clinical.txt" #临床数据文件
setwd("C:\\Users\\Administrator\\Desktop\\geneimmune\\30ROC") #修改工作目录
#读取风险输入文件
risk=read.table(riskFile, header=T, sep="\t", check.names=F, row.names=1)
risk=risk[,c("futime", "fustat", "riskScore")]
#读取临床数据文件
cli=read.table(cliFile, header=T, sep="\t", check.names=F, row.names=1)
#合并数据
samSample=intersect(row.names(risk), row.names(cli))
risk1=risk[samSample,,drop=F]
cli=cli[samSample,,drop=F]
rt=cbind(risk1, cli)
#定义颜色
bioCol=rainbow(ncol(rt)-1, s=0.9, v=0.9)
#绘制ROC曲线
predictTime=3 #定义预测年限
aucText=c()
pdf(file="ROC.pdf", width=6, height=6)
#绘制风险得分的ROC曲线
i=3
ROC_rt=timeROC(T=rt$futime,
delta=rt$fustat,
marker=rt[,i], cause=1,
weighting='aalen',
times=c(predictTime),ROC=TRUE)
plot(ROC_rt, time=predictTime, col=bioCol[i-2], title=FALSE, lwd=2)
aucText=c(paste0("Risk", ", AUC=", sprintf("%.3f",ROC_rt$AUC[2])))
abline(0,1)
#对临床数据进行循环,绘制临床数据的ROC曲线
for(i in 4:ncol(rt)){
ROC_rt=timeROC(T=rt$futime,
delta=rt$fustat,
marker=rt[,i], cause=1,
weighting='aalen',
times=c(predictTime),ROC=TRUE)
plot(ROC_rt, time=predictTime, col=bioCol[i-2], title=FALSE, lwd=2, add=TRUE)
aucText=c(aucText, paste0(colnames(rt)[i],", AUC=",sprintf("%.3f",ROC_rt$AUC[2])))
}
#绘制联合的ROC曲线
multiCox=coxph(Surv(futime, fustat) ~ ., data = rt)
score=predict(multiCox, type="risk", newdata=rt)
ROC_rt=timeROC(T=rt$futime,
delta=rt$fustat,
marker=score,cause=1,
weighting='aalen',
times=c(predictTime),ROC=TRUE)
plot(ROC_rt, time=predictTime, col=bioCol[ncol(rt)-1], title=FALSE, lwd=2, add=TRUE)
aucText=c(aucText, paste0("Risk+Clinical", ", AUC=", sprintf("%.3f",ROC_rt$AUC[2])))
#绘制图例,得到ROC曲线下的面积
legend("bottomright", aucText,lwd=2,bty="n",col=bioCol[1:(ncol(rt)-1)])
dev.off()
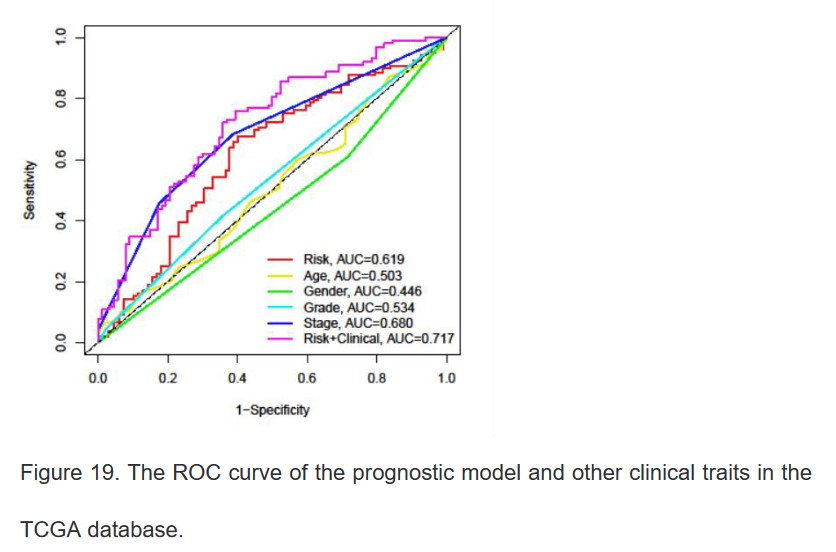
17、绘制列线图与校准曲线
#install.packages("rms")
library(rms) #引用包
riskFile="risk.txt" #风险输入文件
cliFile="clinical.txt" #临床数据文件
setwd("C:\\Users\\Administrator\\Desktop\\生信文章\\geneimmune\\31Nomo") #修改工作目录
#读取风险输入文件
risk=read.table(riskFile, header=T, sep="\t", check.names=F, row.names=1)
risk=risk[,c("futime", "fustat", "riskScore")]
#读取临床数据文件
cli=read.table(cliFile, header=T, sep="\t", check.names=F, row.names=1)
#合并数据
samSample=intersect(row.names(risk), row.names(cli))
risk1=risk[samSample,,drop=F]
cli=cli[samSample,,drop=F]
rt=cbind(risk1, cli)
paste(colnames(rt)[3:ncol(rt)],collapse="+")
#数据打包
dd <- datadist(rt)
options(datadist="dd")
#生成函数
f <- cph(Surv(futime, fustat) ~ riskScore+Age+Gender+Grade+Stage+T+M+N, x=T, y=T, surv=T, data=rt, time.inc=1)
surv <- Survival(f)
#建立nomogram
nom <- nomogram(f, fun=list(function(x) surv(1, x), function(x) surv(2, x), function(x) surv(3, x)),
lp=F, funlabel=c("1-year survival", "2-year survival", "3-year survival"),
maxscale=100,
fun.at=c(0.99, 0.9, 0.8, 0.7, 0.5, 0.3,0.1,0.01))
#nomogram可视化
pdf(file="Nomogram.pdf",height=8.5,width=9.5)
plot(nom)
dev.off()
#calibration curve
time=3 #预测年限
f <- cph(Surv(futime, fustat) ~ riskScore+Age+Gender+Grade+Stage+T+M+N, x=T, y=T, surv=T, data=rt, time.inc=time)
cal <- calibrate(f, cmethod="KM", method="boot", u=time, m=75, B=1000)
pdf(file="calibration.pdf", width=9.5, height=8.5)
plot(cal,
xlab=paste0("Nomogram-Predicted Probability of ", time, "-Year OS"),
ylab=paste0("Actual ", time, "-Year OS(proportion)"),
col="red", sub=T)
dev.off()

18、最后在ICGC肿瘤数据库中再次验证该模型的准确性,代码与以上类似


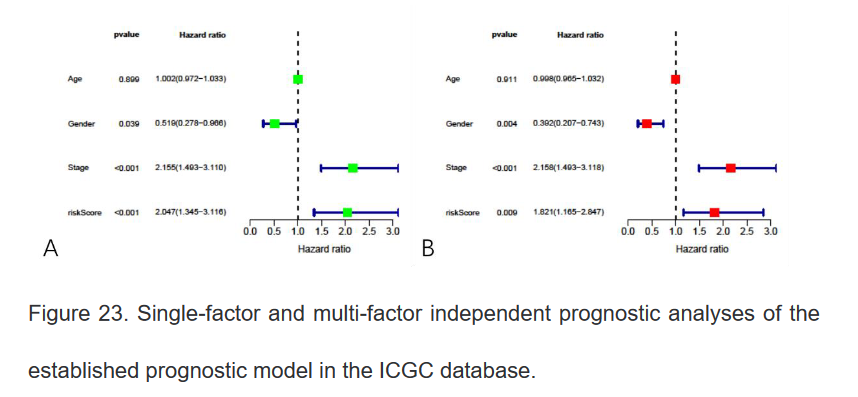

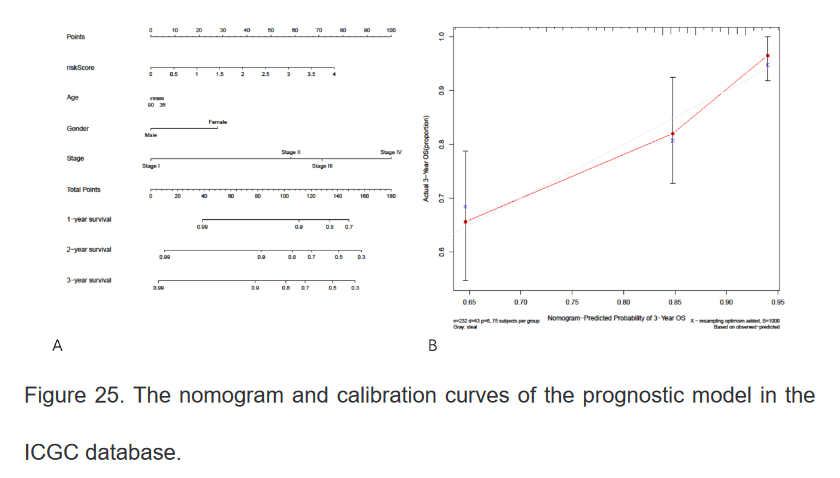
三、总结:
这里只展示了研究的部分内容,有部分研究结果是使用从在线数据库分析获取的,比如中性粒细胞和CD8阳性T细胞的变化情况。研究整体说明的问题是抑制HSP90β可能会通过调节筛选出的免疫基因来改善肝细胞癌患者的预后,后续可以增加部分验证实验来证明研究结果,通过抑制HSP90β来研究不同免疫基因的改变情况。更详细的研究内容可通过以下访问链接获取:
Gitee码云:
Github:
作者:wangyudong
本文版权归作者和博客园共有,欢迎转载,但必须给出原文链接,并保留此段声明,否则保留追究法律责任的权利。


【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· winform 绘制太阳,地球,月球 运作规律
· AI与.NET技术实操系列(五):向量存储与相似性搜索在 .NET 中的实现
· 超详细:普通电脑也行Windows部署deepseek R1训练数据并当服务器共享给他人
· 上周热点回顾(3.3-3.9)
· AI 智能体引爆开源社区「GitHub 热点速览」