

如何基于R包做GO分析?实现秒出图
GO分析
基因本体论(Gene Ontology, GO)是一个用于描述基因和基因产品属性的标准术语体系。它提供了一个有组织的方式来表示基因在生物体内的各种角色。基因本体论通常从三个层面对基因进行描述:细胞成分(Cellular Component,CC)、生物学过程(Biological Process,BP)和分子功能(Molecular Function,MF)。
示例:
细胞成分(Cellular Component,CC):这个层面描述了基因产物(如蛋白质)在细胞内的定位。例如,它们可能位于细胞核、细胞质、线粒体膜或其他细胞器上。这有助于了解基因产物在细胞内的作用和功能。
生物学过程(Biological Process,BP):这个层面描述了基因参与的生物学过程。这些过程可能包括细胞生长、信号传导、基因表达调控、代谢途径等。通过了解基因参与的生物学过程,我们可以更好地理解生物体的生理功能和疾病发生机制。
分子功能(Molecular Function,MF):这个层面描述了基因产物在分子层面的功能,通常涉及到与其他分子的相互作用或催化生化反应。例如,某个基因产物可能是一个酶,它能够催化某个特定的生化反应;或者它可能是一个结构蛋白,参与细胞骨架的组装和维持。
本期将介绍使用R clusterProfiler包对基因进行GO功能富集分析、OrgDb包制作以及结果可视化。使用R clusterProfiler包对基因进行富集,需要导入目的基因(前景基因)相对应物种的参考基因组(背景基因),现阶段“Bioconductor”已有十几种常见动物,如人类、小鼠等物种的OrgDb。但仍然有许多物种不在Bioconductor的OrgDb列表里,但存在参考基因组,如山羊,绵羊等,因此大家可以选择性使用。
上篇文章我们已经用Perl脚本将蛋白名批量转化为了基因名,下面首先用R语言将基因名找到基因ID,以作后续分析。
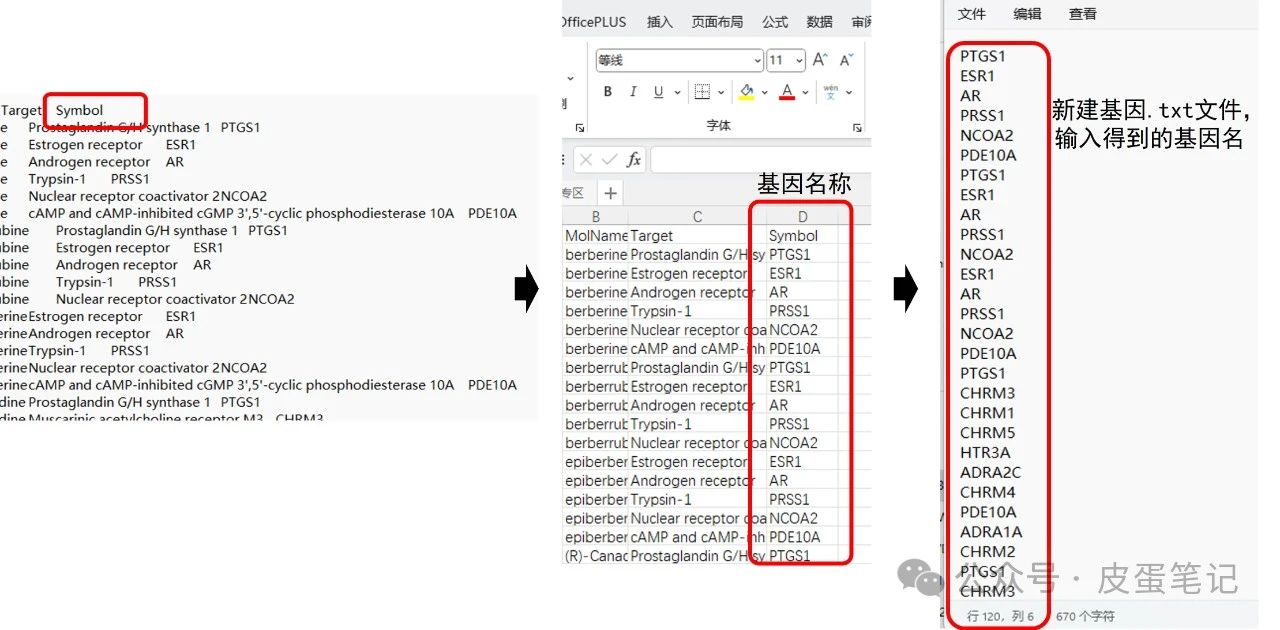
1.在上篇文章转换的结果targetSymbol文件中提取基因名,如图所示,建立“基因.txt”文件
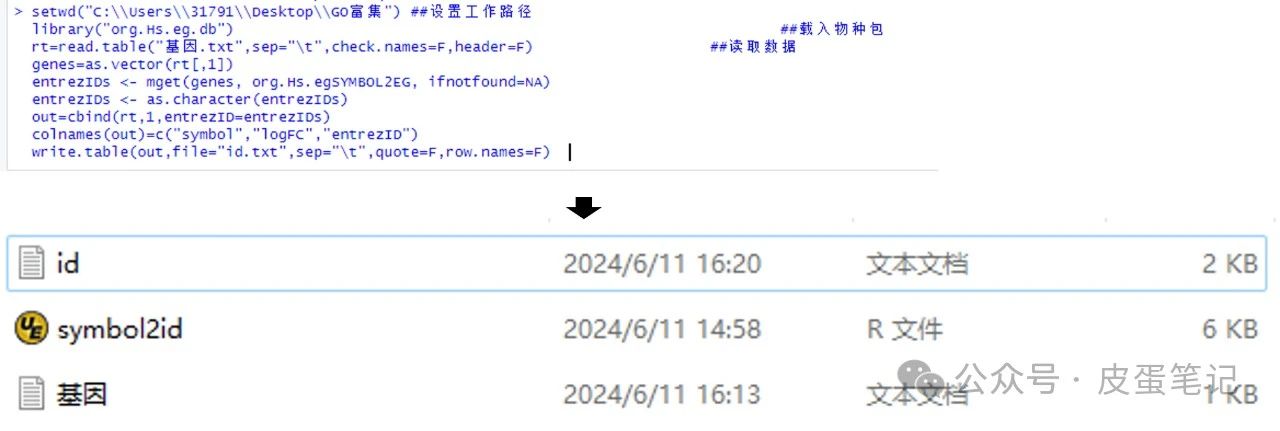
2.导入文件,运行代码,得到id.txt文件
library("org.Hs.eg.db") ##载入物种包
##id转换
rt=read.table("基因.txt",sep="\t",check.names=F,header=F) ##读取数据
genes=as.vector(rt[,1])
entrezIDs <- mget(genes, org.Hs.egSYMBOL2EG, ifnotfound=NA)
entrezIDs <- as.character(entrezIDs)
out=cbind(rt,1,entrezID=entrezIDs)
colnames(out)=c("symbol","logFC","entrezID")
write.table(out,file="id.txt",sep="\t",quote=F,row.names=F)
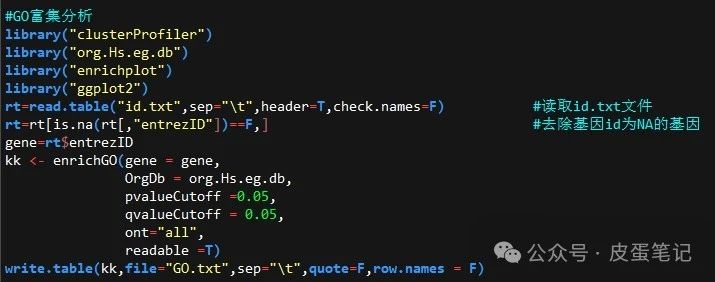
3.GO分析,将上述分析得到的id.txt文件导入,运行以下代码以做GO分析
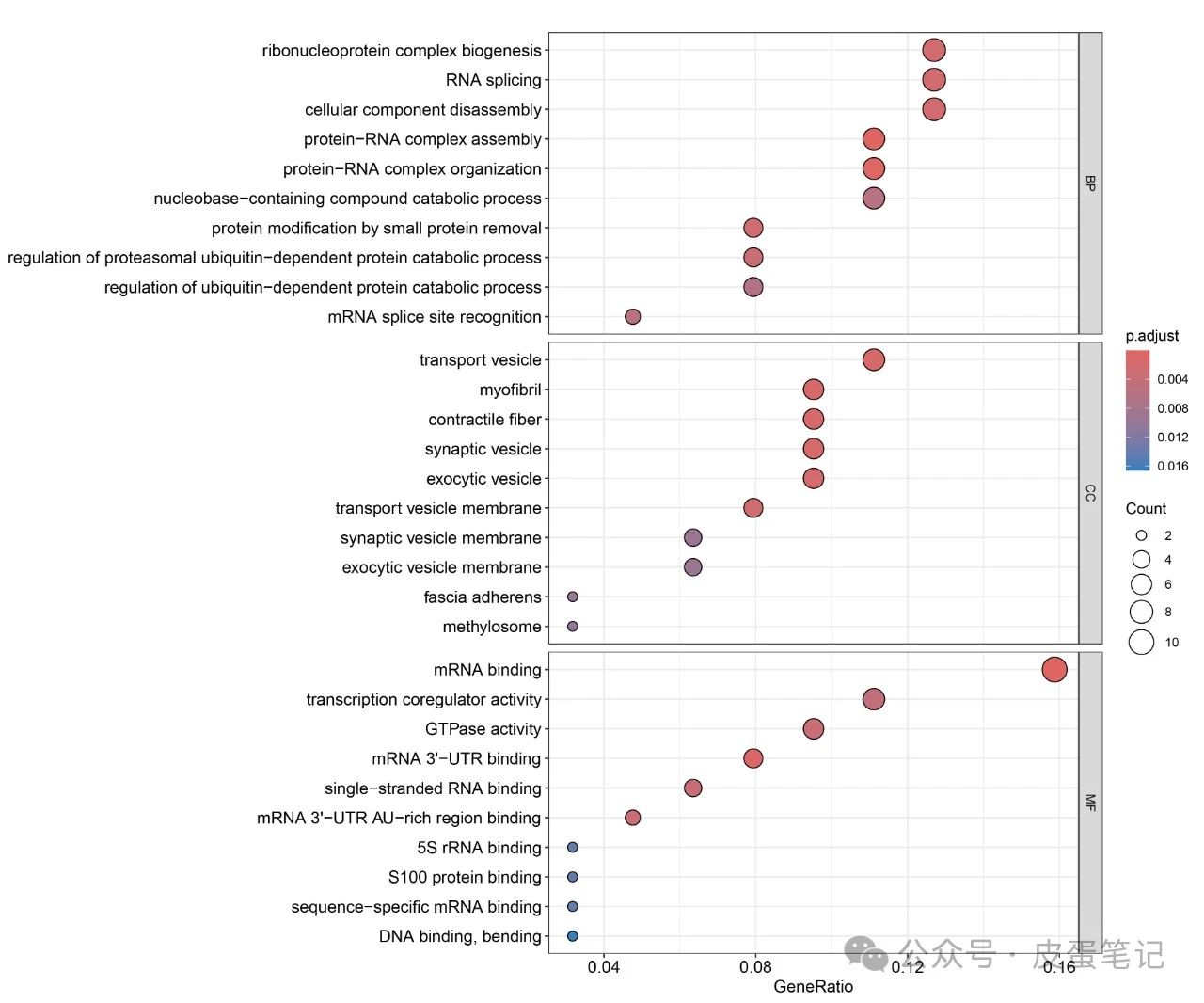
4.得到GO文件,绘制气泡图和柱状图
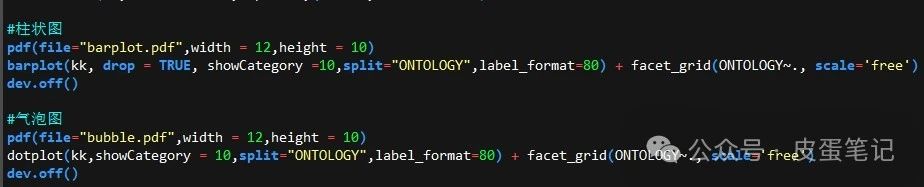
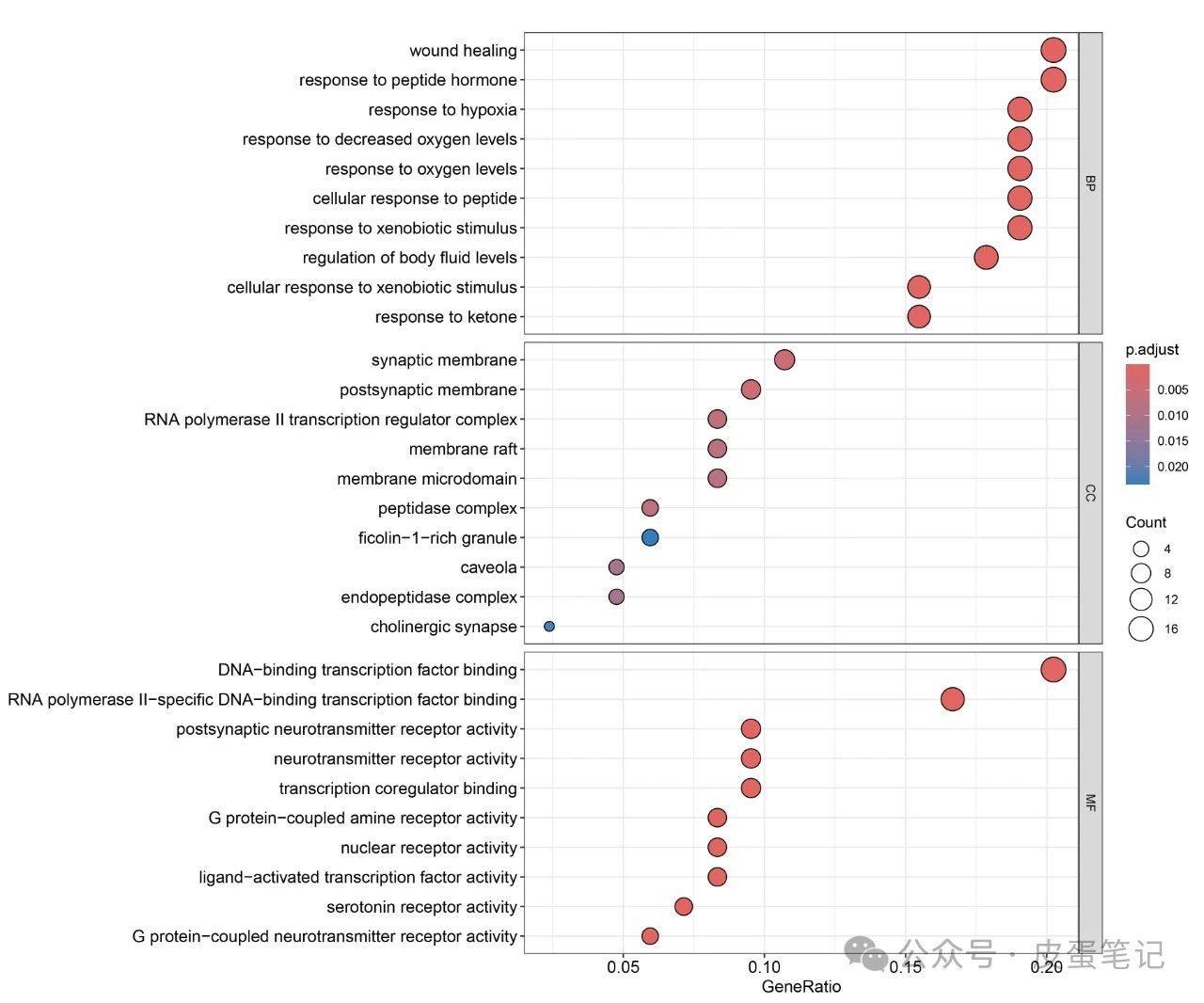
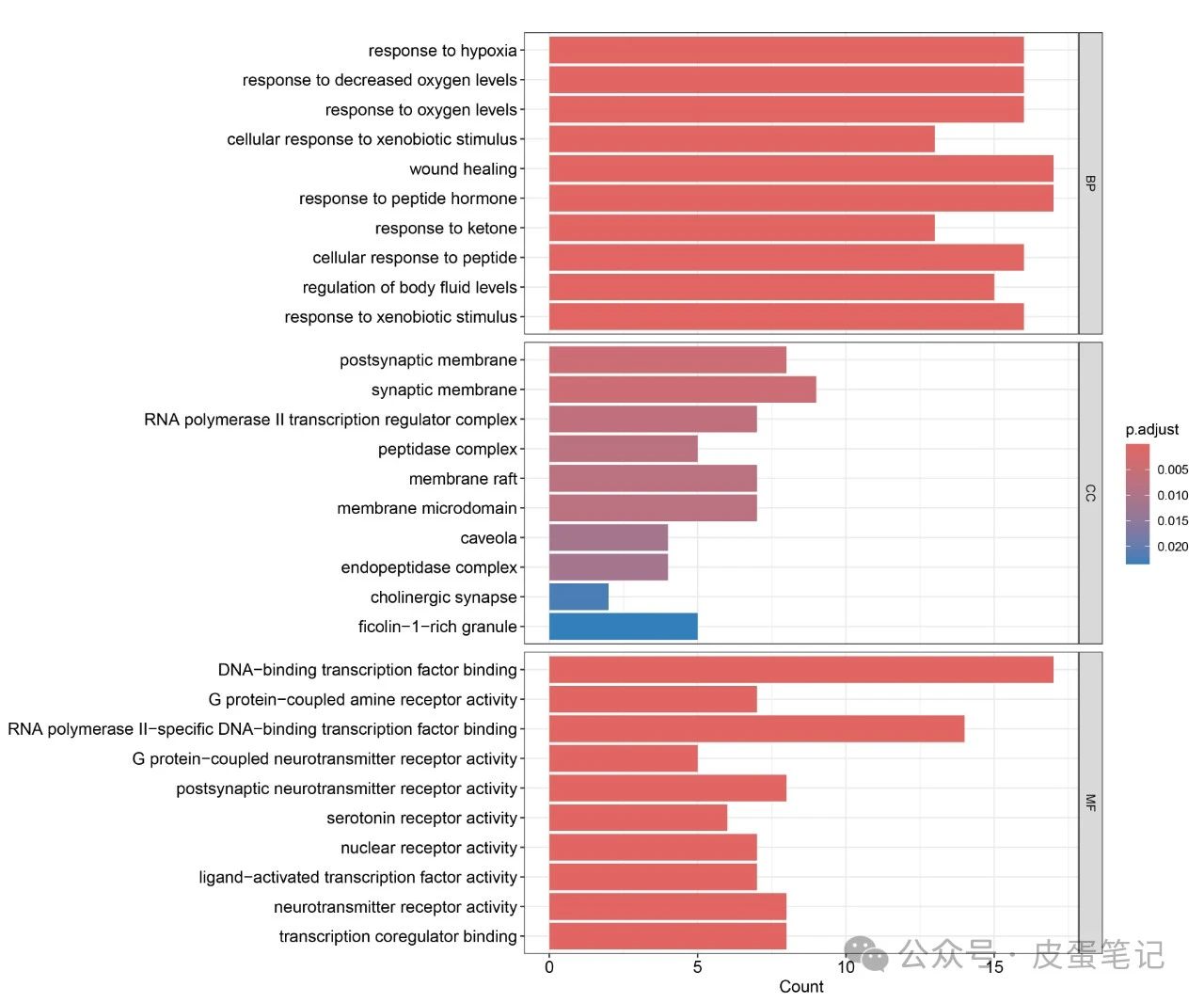
下篇笔记将会展示如何应用R语言作KEGG分析,敬请关注与期待。整理不易用你们发正刊(CNS)的金手指为本篇内容点赞支持,本文代码与文件在以下链接。
链接:https://pan.baidu.com/s/1CW6Trynp9DNPhyCncFBLfA
提取码:0jsj
文章转载自公众号:皮蛋笔记,欢迎关注,随时获取第一手文章内容。

【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步