

如何基于Perl实现批量蛋白名转换为基因名?以做后续GO与KEGG分析
众所周知,在完成蛋白组学组间差异蛋白筛选后,往往要做GO与KEGG功能富集分析,这就需要我们首先将蛋白名转换为基因名,或者找出基因ID。将蛋白名转化为基因名可能涉及不同的转换工具或数据库,这里有几种常见的方法:
①UniProt数据库:UniProt数据库提供了蛋白和其对应基因的关联信息。可以通过查询UniProt数据库来找到蛋白名对应的基因名。
② 基因注释工具:一些基因注释工具(如DAVID、Enrichr等)能够接受蛋白名作为输入,并返回与之关联的基因信息。
③文献和数据库搜索:通过文献或特定的生物信息学数据库(如NCBI、Ensembl等),可以手动查找蛋白名和基因名之间的关联。
④转录本和基因组数据库:有时蛋白和基因之间的映射可以通过转录本和基因组序列的对应关系来确定。
以上四种方法虽然可以实现蛋白名与基因名的转换,但是只适应于小样本的处理,如果差异有几百或者上千个蛋白,那么用以上方法处理起来会非常麻烦。下面我们将介绍如何用perl进行批量处理。源代码可关注本公众号(皮蛋笔记)私信获取。
1、首先下载并安装Perl脚本
网页搜索Perl直接进入即可(Strawberry Perl for Windows),点击下载并安装。

2、准备所需文件
Protein name.txt为筛选到的差异蛋白文件如下图所示,包含蛋白名称或者蛋白ID,本例中所使用的为蛋白名称。uniprot_Human.tsv为从uniprot数据库下载的物种蛋白数据库,如何下载之前笔记已经讲述。addSymbol文件就是Perl脚本,可关注本公众号(皮蛋笔记)私信获取。

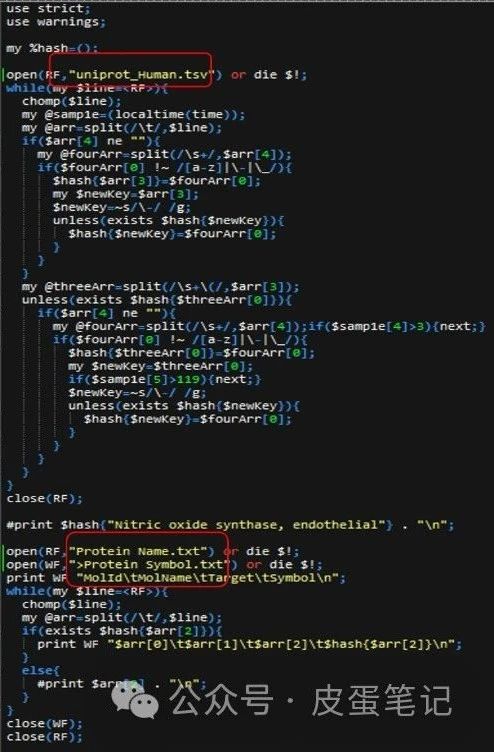
3.打开addSymbol文件,更改代码中的文件名,以输入、输出文件
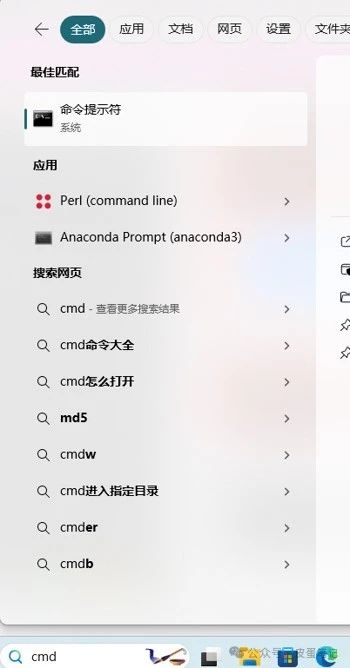
4.在电脑搜索框中输入“cmd”,打开命令提示符。
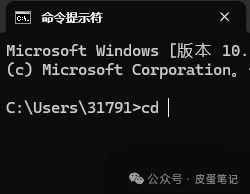
5.如下图所示,输入“cd ”(cd后有空格),将文件所在工作路径复制或粘贴至cd后面,并按下“enter”打开工作路径
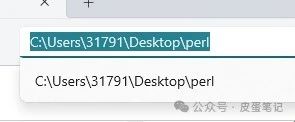

6.手动输入addSymbol.pl文件名,按下“enter”键运行即可得结果。
7.可以发现在文件夹里已经多了结果文件,打开进行后处理即可
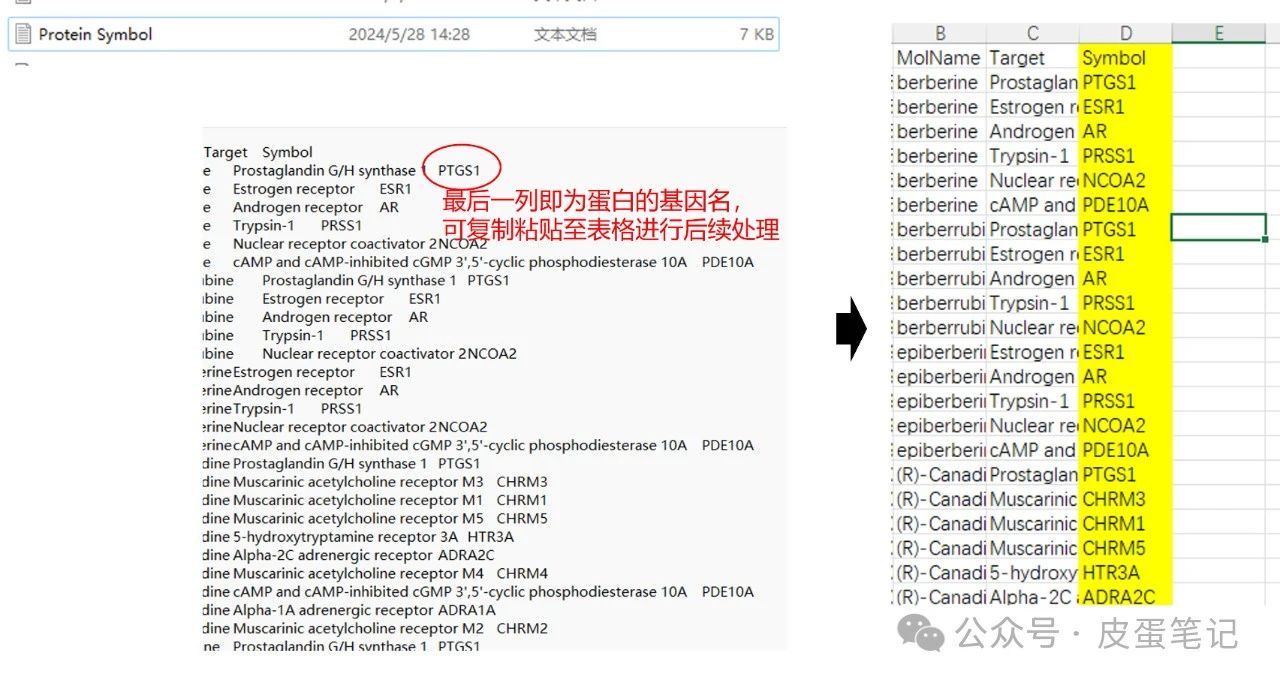
在获取蛋白质的基因的名称后,需要获取基因的ID以为后续进行GO和KEGG分析,下篇笔记将会展示如何应用R语言进行GO分析,敬请关注与期待。本篇内容代码与文件见以下链接
链接:https://pan.baidu.com/s/1oQ9dqOeHdO5I08skBt40VQ
提取码:r3ml
内容转自公众号:皮蛋笔记,欢迎关注,获取第一时间咨询和相关资料。



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· winform 绘制太阳,地球,月球 运作规律
· 超详细:普通电脑也行Windows部署deepseek R1训练数据并当服务器共享给他人
· 上周热点回顾(3.3-3.9)
· AI 智能体引爆开源社区「GitHub 热点速览」
· 写一个简单的SQL生成工具