如何通过SRA Tools处理从NCBI获得的SRA数据
1.安装SRA Tools
通过SRA Toolkit可以方便的从NCBI下载SRA数据,但是速度较慢,Aspera虽然快,但是难点在于找NCBI的源文件地址,而且SRA Toolkit好像可以调用Aspera(虽然还没找到方法)
具体操作可以参考这个帖子,下载安装很容易,主要是配置环境要配置好,不然用不了
https://blog.csdn.net/m0_69574256/article/details/134645370
2.下载SRA数据
从NCBI网站上进SAR,找到自己感兴趣的数据

选择sent to
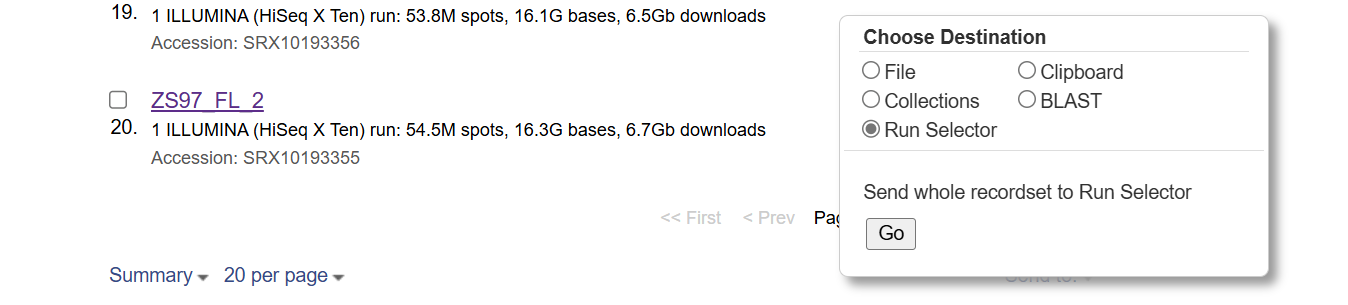
然后选择Run selector,然后GO,他会输出一个SRR_Acc_List.txt,这个文件里面包含了你想要的数据的编号,之后可以通过这个文件批量下载,或者通过里面的编号一个一个下,里面的内容都是SRRxxxxxx

想要下载这些数据时,可以参考这个帖子
https://blog.csdn.net/m0_69574256/article/details/134645370
主要操作就是:
prefetch SRRXXXXXX(这里是你想要下的数据的标号),这个命令是下载这个SAR文件
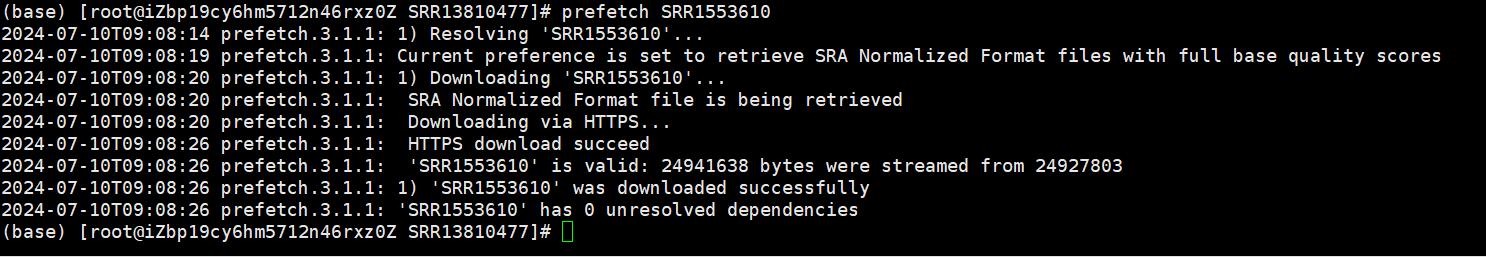
这样就是下载好了
对于下载好的SRA文件来说,就下来就是提取数据了,使用如下命令可以提取数据:
fastq-dump --split-3 --gzip SRRxxxxxxx.sra
fastq-dump:提取内容的命令,提取fastq, fastq-dump这步很容易出错,一定要注意原始数据的类型是单端测序还是双端测序,单端测序和双端测序处理方法不同
--split-3:这个要着重说一下,因为单端测序和双端测序是不一样的, 双端测序不要直接提取,将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件夹里
关于遇到的Rejected XXXXX READS because of filtering out non-biological READS就是因为原来是SE数据,但是用--split-3当作PE数据处理,出现的问题. 看起来好像有问题,但是对后续结果分析没有太多影响.
因此,对于一个你不知道到底是单端还是双端的SRA文件,一律用--split-3.
--gzip:将提取出来的fastq文件转换为gz文件,可以节省服务器存储资源,同时gz文件也便于后续处理
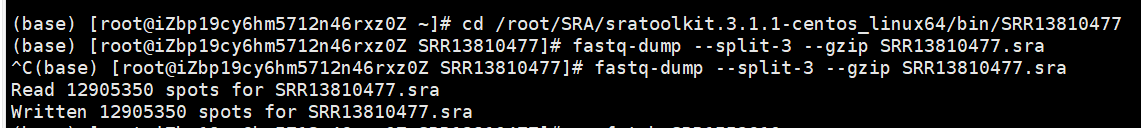
下面就是数据提取出来的最终形态,看得出来这个对存储大小要求很高,不压缩会很占空间,记得在下载和提取过程中留足充足的磁盘资源,不然会报错,内存越大越好,这样提取的更快。



【推荐】国内首个AI IDE,深度理解中文开发场景,立即下载体验Trae
【推荐】编程新体验,更懂你的AI,立即体验豆包MarsCode编程助手
【推荐】抖音旗下AI助手豆包,你的智能百科全书,全免费不限次数
【推荐】轻量又高性能的 SSH 工具 IShell:AI 加持,快人一步
· 分享一个免费、快速、无限量使用的满血 DeepSeek R1 模型,支持深度思考和联网搜索!
· 基于 Docker 搭建 FRP 内网穿透开源项目(很简单哒)
· ollama系列1:轻松3步本地部署deepseek,普通电脑可用
· 按钮权限的设计及实现
· Apache Tomcat RCE漏洞复现(CVE-2025-24813)