“屎”上最大:基于粪便微生物组的疾病诊断模型
前言
人类的肠道中存在着大量的微生物,这些微生物在消化、代谢甚至疾病预防方面都发挥着重要作用,而肠道微生物群一旦失衡,就有可能导致一些疾病的发生。研究表明健康人的肠道微生物群可以反映一些疾病的特征,因此可以当作疾病诊断的参考。最近,来自香港中文大学的一个团队将机器学习的研究方法应用到了香港华人的粪便样本宏基因组测序数据,得到了目前为止涵盖多种疾病的最大单点数据集,并且基于此数据建立了粪便微生物组的疾病预测模型并在不同人群中进行了验证。

数据的收集与测序
作者对来自2320名香港华人的粪便样本进行了宏基因组测序,这些样本提供者包含了9种明确的疾病表型,分别是结直肠癌(CRC, n=174)、结直肠腺瘤(CA, n=168)、克罗恩病(CD, n=200)、溃疡性结肠炎(UC, n=147)、肠易激综合征-腹泻亚型(IBS-D, n=145)、肥胖(n=148)、心血管疾病(CVD, n=143)、记性COVID-19综合征(PACS, n=302)以及893名健康对照。

测序后,作者一共获得了14.3 TB的测序数据,一共鉴定到了1208个细菌物种,其中,325种细菌的相对丰度高于0.15%,这些物种存在于超过5%的受试者中。
不同疾病之间的共同微生物组特征
作者观察到,在不同疾病中,细菌多样性和物种丰富度存在着差异,这个差异与另外一项荟萃研究的结果一致,表明这些生态指标可能不是区分健康或患病的可靠指标。随后,作者使用MaAsLin2的线性模型分析了物种水平上的微生物组成与疾病的关联,发现与疾病关联的细菌物种中的大多数都与不止一种疾病相关,说明不同的疾病往往有着很多共同的特征。作者又尝试了两种二元分类器,发现二元分类器不能仅仅根据单种疾病的数据准确预测疾病的特异性特征。于是,作者便尝试了基于多分类器的方法。
基于粪便微生物组的多分类模型开发
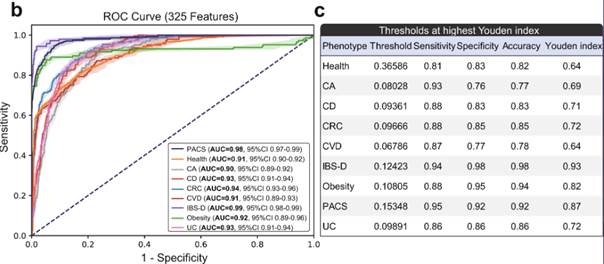
作者尝试了5种多分类模型(图1中间的5种模型),分别是RF、KNN、MLP、SVM和GCN,并测试了这些模型的效果。结果显示,这物种模型的性能都高于二元分类模型,表明多分类模型更加适合用来从粪便微生物组数据中预测疾病。RF模型在这些模型中具有最好的性能(0.90~0.99),因此,作者设计了很多测试实验来评估RF模型的性能,结果都显示RF模型具有不错的表现。
在独立数据集上的验证
证明了模型在训练数据中的良好表现后,自然就要证明模型在其他数据上也有不错的表现。作者选择了另一个涵盖了3个地区的宏基因组数据集对模型进行了测试,作者的RF模型获得了0.69~0.91的分类准确率,再次说明了模型的稳健性。为了进一步验证模型的准确性,作者选择了60名新冠痊愈者(没有后遗症的痊愈者)进行分类预测,结果显示RF模型将这些受试者分类为健康人的准确率为83.3%,这说明,新冠痊愈者的肠道微生物组与健康人相似。然而,作者检查了不在研究中的两种疾病的分类表现,发现RF模型对于其他疾病的预测效果较低。
将菌群特征作为疾病预测标志物
作者将模型中贡献最大的前50个细菌物种与疾病表型进行了关联来检查这些物种对模型的贡献。结果显示几乎所有的疾病表型都与厚壁菌或放线菌门微生物的低丰度相关,而与拟杆菌的高丰度相关。厚壁菌与拟杆菌的比例失衡曾被报道在肥胖和IBD中出现,但其他的情况还没有被报道过,说明这可能是一种潜在的致病因素。最后,作者根据关联的结果,总结了各种疾病中可能存在的细菌失衡类型。
总结
这项研究的亮点有两个,其一是高质量的粪便微生物组的宏基因组测序,其二是不同机器学习方法的对比以及经过各种验证的方法。这项研究为肠道微生物组与疾病的关联研究提供了一个新的思路,并展示了机器学习方法在这类研究中的应用。
参考文献
Su, Q., Liu, Q., Lau, R.I. et al. Faecal microbiome-based machine learning for multi-class disease diagnosis. Nat Commun 13, 6818 (2022). https://doi.org/10.1038/s41467-022-34405-3
本文来自博客园,作者:冻羊,转载请注明原文链接:https://www.cnblogs.com/wdyyy/p/16950400.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号