三代测序--QC篇
之前总结的有关Pacbio RSII测序数据QC的一些基本知识。
Part 1. 下机数据
用一个下机数据作为示例:

在analysis文件夹中,下机的数据被分割为三个文件进行存储,其中以bax.h5为后缀的是原始二进制文件;以subreads.fasta / subreads.fastq为后缀的是经一级处理得到的标准格式的碱基文件;以sts.csv / sts.xml为后缀的是记录测序过程中每个ZMW度量指标的统计文件。
数据的命名:
m140415_143853_42175_c100635972550000001823121909121417_s1_p0
└1┘└───2───┘└─3─┘└────────4───────────────┘└5┘└6┘
1. m是movie的缩写;
2. 测序时间,格式为yymmdd_hhmmss;
3. 仪器编号;
4. SMRT Cell Barcode;
5和6无实际意义,一般是固定的。
Part 2. 数据结构
要做好数据的质控,不仅要知其然,还有知其所以然。
Pacbio数据的文库模型是两端加接头的哑铃型结构,测序时会环绕着文库进行持续的进行,由此得到的测序片段称为polymerase reads,即一条含接头的测序序列,其长度由反应酶的活性和上机时间决定。目前,采用最新的P6-C4酶,最长的读长可达到60kb以上。
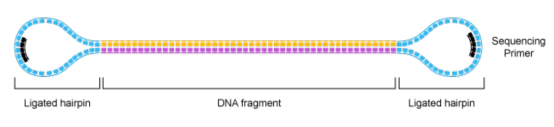
polymerase reads是需要进行一定的处理才能获得用于后续分析的。这个过程首先是去除低质量序列和接头序列:

处理后得到的序列称为subreads,根据不同文库的插入片段长度,subreads的类型也有所不同。
在用于基因组denovo时,通常会构建10kb/20kb的文库,对长插入片段文库的测序基本是少于2 passes的(pass即环绕测序的次数),得到的reads也称为Continuous Long Reads (CLR),这样的reads测序错误率等同于原始的测序错误率。
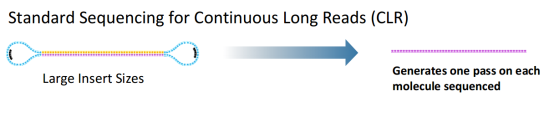
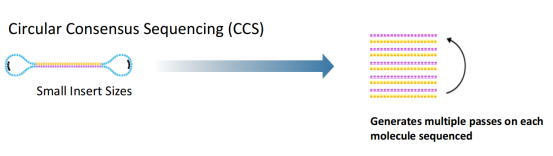
而对于全长转录组或全长16s测序,构建的文库插入片段较短,测序会产生多个passes,这时会对多个reads进行一致性校正,得到一个唯一的read,也称为Circular Consensus Sequencing(CCS)Reads,这样的reads测序准确率会有显著的提升。
Part 3. 数据质量
不同于二代测序的碱基质量标准Q20/Q30,三代测序由于其随机分布的碱基错误率,其单碱基的准确性不能直接用于衡量数据质量。那么,怎么判断三代测序的数据好不好呢?
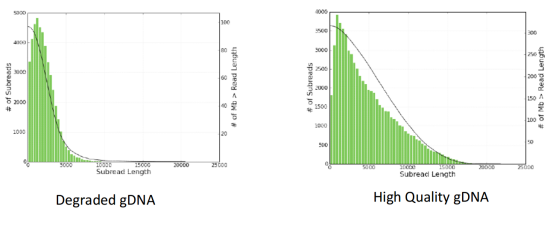
最直接的方法是看长度。长度短的测序数据不一定差(与文库大小有关),但差的数据长度一定短。在上游实验环节,最关键的影响因素是文库的构建。高质量的文库产出的数据长度长,质量好;而低质量的文库产出的数据长度短,质量差。
其次,看比例。需要关注的是两个比例,一个是subreads与polymerase reads数据量的比例,比例过低反映测序过程中的低质量的序列较多;一个是zmw孔载入的比例,根据孔中载入的DNA片段数分为P0、P1和P2。P1合理比例在40%-60%之间。上样浓度异常会导致P0或P2比例过高,有效数据量减少。需要注意的是P2比例过低时,可能存在P2转P1的情况,测序结果包含较多的嵌合型reads。
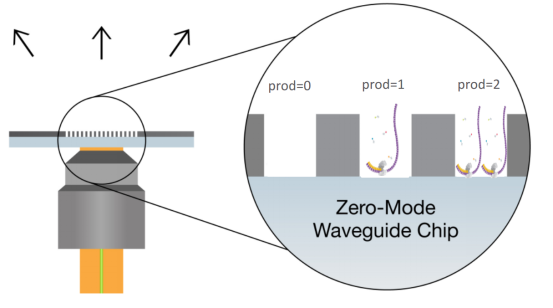
现在Sequel测序仪器已经稳定且通量更高,不过相应的QC思路大体类似,具体的差异本篇暂不讨论,待后续更新。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号