【变异检测】CNV PAV INV TRANS变异检测
前言
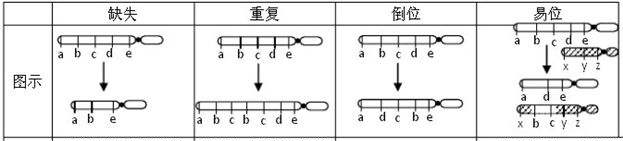
SV(大片段结构变异)指在基因组水平上大片段的insertion,deletion,inversion,translocation,duplication等变异。
SV检测分重测序部分和基因组部分,重测序又分二代测序数据和三代测序数据之分。每种分析方法用到的软件是不一样的,但结果可能有重叠部分。
针对于基因组,检测出的SV的准确度主要取决于基因组组装质量,三代组装的结果更优于二代组装,以及基因组对比软件的检出的准确性。SV检测原理大致就是两个基因组之间在某些片段是高度相似,比对上后针对于这些片段进行找对应位置是否有对应高度一致的序列。如果相对于参考基因组片段,比对的基因组没有这段序列则为deletion变异;而相对于比对的基因组片段,参考基因没有则为insersion变异。如果参考基因组与比对基因组在高度相似片段序列是处于翻转的,则为inversion变异。
如果参考基因组与比对基因组在高度相似片段不是染色体位置对应附近,距离相隔比较远,则为translocation变异。如果参考基因组与比对基因组上高度相似片段在其他位置也有,那为duplication变异。其中inversion、translocation变异还需要侧翼序列一起来判定。
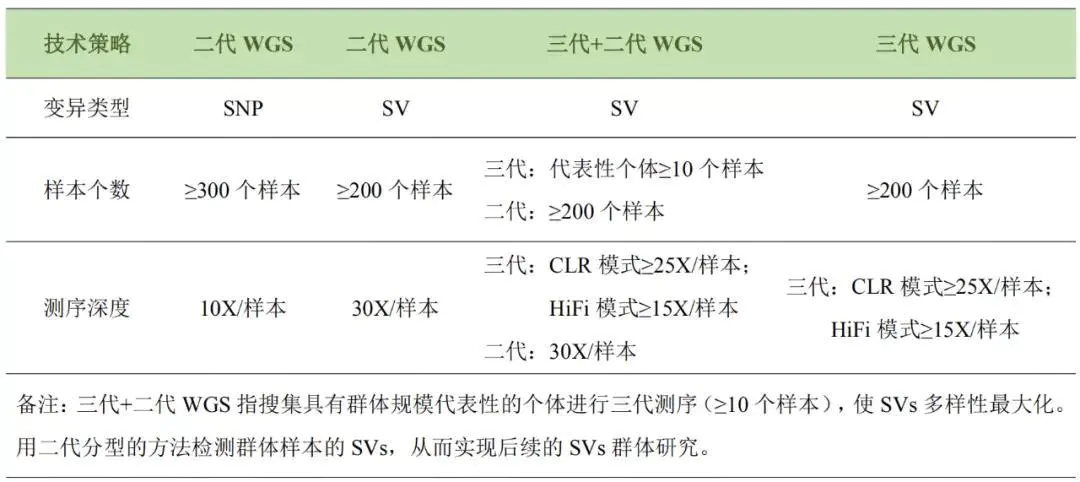
针对重测序,包括二代和三代的SV变异检测与基因组差不多,除了使用软件不一样,最大不同是序列长度和只比对参考基因组上(非相互比对,仅单一比对到参考基因组),所以导致变异种类和数量会少一些,比如insertion可能检测不到。
注:重测序SV检测需要一定测序深度支持
分析内容
1.二代
二代重测序数据检测SV主要用这两个软件lumpy-sv和delly软件
Lumpy-sv软件下载和安装:GitHub - arq5x/lumpy-sv: lumpy: a general probabilistic framework for structural variant discovery
git clone --recursive https://github.com/arq5x/lumpy-sv.git
cd lumpy-sv
make
cp bin/* /usr/local/bin/
delly软件下载和安装:GitHub - dellytools/delly: DELLY2: Structural variant discovery by integrated paired-end and split-read analysis
git clone --recursive https://github.com/dellytools/delly.git
cd delly/
make all
Delly软件是专门SV预测方法,可以在short-reads中大规模基因分型和可视化的
串联重复、倒位、易位、缺失等变异。它使用paired-ends、split-reads、read-depth来准确地描绘整个基因组中的基因组重排。结构变体可以使用delly-sansa进行注释,并使用delly-maze或delly-suave进行可视化。
###
b=`cat rmdup.bam.list|tr "\n" ","`
b=${b%,}
s=`cat splitters.bam.list|tr "\n" ","`
s=${b%,}
d=`cat discordants.bam.list|tr "\n" ","`
d=${b%,}
lumpyexpress -B $b -S $s -D $d -o test_lumpy-sv.vcf
#
rmdup.bam.list文件格式
splitters.bam.list文件格式
discordants.bam.list文件格式
#extractSplitReads_BwaMem为lumpy软件中一个程序
samtools view -h test.rmdup.bam | extractSplitReads_BwaMem -i stdin | samtools view -Sb - >test.splitters.bam
samtools view -b -F 1294 test.rmdup.bam >test.discordants.bam
合并多个方法检测出来的SV,可以用SURVIVOR软件(三代测序中也有描述)
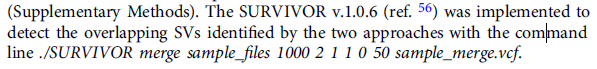
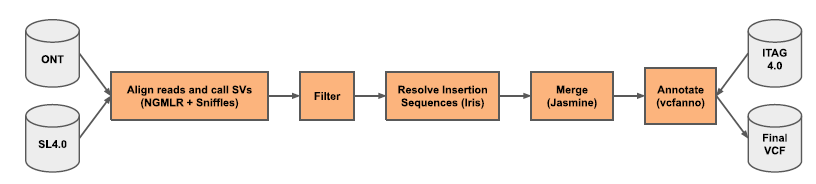
2.三代long-reads


3.基因组
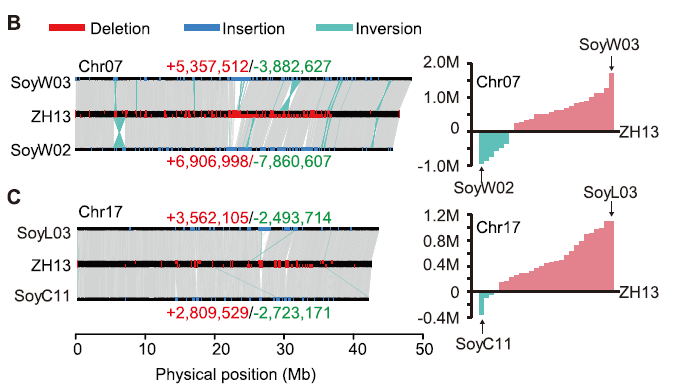
2021cell水稻,2020cell大豆,2018science人
基因组检测SV方法很多,主要是比对软件差异,导致分析方法的差异。比对软件有lastz,minimap2,mummer等,每种软件原理不一,详细去看软件说明。主要讲mummer比对,因为mummer比对最近比较流行的比对软件,且基于比对的结果开发好几种检测SV的软件,比如SVMU,SYRI等
3.1 mummer软件
软件下载:GitHub - mummer4/mummer: Mummer alignment tool
程序1:nucmer
nucmer是基于fasta数据文件之间进行比对的。它最适用于可能有大量重排的相似序列。比如:比较两个基因组组装,将组装或测序reads mapping到已经完成的基因组,以及比较可能有大量重排和重复的相关物种的两个基因组。
USAGE:
nucmer [options] <reference> <query>
[options] type 'nucmer -h' for a list of options.
<reference> specifies the multi-FastA sequence file that contains
the reference sequences, to be aligned with the queries.
<query> specifies the multi-FastA sequence file that contains
the query sequences, to be aligned with the references.
OUTPUT:
out.delta the delta encoded alignments between the reference and
query sequences. This file can be parsed with any of
the show-* programs which are described in the "RUNNING
THE MUMmer UTILITIES" section.
nucmer -c 1000 --mum --maxgap=1000 --prefix=ref_qry --threads=20 ref.fa qry.fa
程序2:dnadiff
dnadiff是基于nucmer的结果,输出是的比对统计、snp、断点。它主要是评估两个高度结构相似序列的相似集。
USAGE: dnadiff [options] <reference> <query>
or dnadiff [options] -d <delta file>
<reference> Set the input reference multi-FASTA filename
<query> Set the input query multi-FASTA filename
or
<delta file> Unfiltered .delta alignment file from nucmer
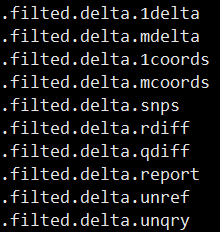
OUTPUT:
.report - Summary of alignments, differences and SNPs
.delta - Standard nucmer alignment output
.1delta - 1-to-1 alignment from delta-filter -1
.mdelta - M-to-M alignment from delta-filter -m
.1coords - 1-to-1 coordinates from show-coords -THrcl .1delta
.mcoords - M-to-M coordinates from show-coords -THrcl .mdelta
.snps - SNPs from show-snps -rlTHC .1delta
.rdiff - Classified ref breakpoints from show-diff -rH .mdelta
.qdiff - Classified qry breakpoints from show-diff -qH .mdelta
.unref - Unaligned reference IDs and lengths (if applicable)
.unqry - Unaligned query IDs and lengths (if applicable)
dnadiff -d ref_qry.filted.delta -p ref_qry.filted.delta
程序3:delta-filter
该程序过滤由nucmer或promer生成的delta对齐文件,只留下所需要的对齐,这些对齐以与输入相同的delta格式输出到stdout。它的主要功能是LIS算法,它计算最长递增的比对子集。这允许使用-g选项或局部一致的-1或-m计算一组全局对齐(即1对1和相互一致的顺序)。可以使用-r将参考序列映射到查询序列,或使用-q将查询映射到参考。这允许用户排队机会并重复诱导比对,只留下两个数据集之间的“最佳”比对。
USAGE:
delta-filter [options] <deltafile>
[options] type 'delta-filter -h' for a list of options.
<deltafile> the .delta output file from either nucmer or promer.
OUTPUT:
stdout The same delta alignment format as output by nucmer and promer.
delta-filter -i 90 -l 1000 -1 ref_qry.delta > ref_qry.filted.delta
程序4:show-coords
该程序解析nucmer和promer的delta对齐输出,并显示坐标以及有关对齐的其他有用信息。-c和-l选项在比较两组组装contig时很有用,因为这些选项有助于确定对齐是否跨越整个contig,或者只是对不同读取的部分全中。当我们希望识别两个基因组之间的共线区域,但对实际的比对相似性或外观并不特别感兴趣时,-b选项很有用。
USAGE:
show-coords [options] <deltafile>
[options] type 'show-coords -h' for a list of options.
<deltafile> the .delta output file from either nucmer or promer.
OUTPUT:
stdout run 'show-coords' without the -H option to see the column
header tags. Here is a description of each tag. Note that
some of the below tags do not apply to nucmer data, and that
all coordinates are inclusive and relative to the forward DNA
strand.
[S1] Start of the alignment region in the reference sequence.
[E1] End of the alignment region in the reference sequence.
[S2] Start of the alignment region in the query sequence.
[E2] End of the alignment region in the query sequence.
[LEN 1] Length of the alignment region in the reference sequence,
measured in nucleotides.
[LEN 2] Length of the alignment region in the query sequence, measured
in nucleotides.
[% IDY] Percent identity of the alignment, calculated as the
(number of exact matches) / ([LEN 1] + insertions in the query).
[% SIM] Percent similarity of the alignment, calculated like the above
value, but counting positive BLOSUM matrix scores instead of exact
matches.
[% STP] Percent of stop codons of the alignment, calculated as
(number of stop codons) / (([LEN 1] + insertions in the query) * 2).
[LEN R] Length of the reference sequence.
[LEN Q] Length of the query sequence.
[COV R] Percent coverage of the alignment on the reference sequence,
calculated as [LEN 1] / [LEN R].
[COV Q] Percent coverage of the alignment on the query sequence,
calculated as [LEN 2] / [LEN Q].
[FRM] Reading frame for the reference sequence and the reading frame
for the query sequence respectively. This is one of the columns
absent from the nucmer data, however, match direction can easily be
determined by the start and end coordinates.
[TAGS] The reference FastA ID and the query FastA ID.
There is also an optional final column (turned on with the -w
or -o option) that will contain some 'annotations'. The -o option will
annotate alignments that represent overlaps between two sequences,
while the -w option is antiquated and should no longer be used.
Sometimes, nucmer or promer will extend adjacent clusters past one
another, thus causing a somewhat redundant output, this option will
notify users of such rare occurrences.
show-coords -THrd ref_qry.filted.delta >ref_qry.filted.delta.coord
最终得到结果会有以下格式文件:
3.2 SVMU软件
软件下载:GitHub - mahulchak/svmu: A program to call variants from genome alignment
该软件可以用lastz结果,也可以用mummer结果进行检测PAV和CNV。SVMU全名是Structural Variants from MUmmer。
svmu sam2ref.mm.delta ref.fasta sample.fasta snp_mode sam_lastz.txt prefix
其中
snp_mode有两种选择 'h'或者'l'。h = report SNPs; l = no SNPs
prefix为输出文件的前缀.
sv.prefix.txt为一个制表符分隔的文件,汇总了比对基因组相对于参考基因组的SV(PAV,CNV,INV)
small.prefix.txt为一个制表符分隔的文件,包含SNP和小片段的INDEL变异
cnv_all.prefix.txt为一个制表符分隔的文件,其中包含在比对基因组中以较高拷贝数(>1)存在的所有参考基因组区域。名称中带有“trans”代表它是转座子或不同染色体中基因的非TE拷贝
cm.prefix.txt包含两个基因组之间共线的参考基因组区域的文件
svmu ref_qry.filted.delta ref.fa qry.fa h null ref_qry
3.3 syri软件
下载路径:GitHub - schneebergerlab/syri: Synteny and Rearrangement Identifier
syri软件主要是基于mummer的结果开发的一套检测染色体级别变异的工具,变异包含SNP、INDEL、PAV、INV、TRANS等。
SyRI是从识别最长的共线性区域开始的,而对于所有非共线性区域都是通过对应在两个基因组之间共线区域来判定重排,因此鉴定共线区域也会同时鉴定了所有结构重排。以此为基础识别所有SR,将SR分类成各种变异,如倒位,易位或重复。此外,syri还可以识别所有共线性区域和重排区域内的局部变异。Local variations 包括小变异(如SNP和小indel),structural variations(如大indel,CNV(拷贝数变异)和HDR)。从比对结果中解析出短的变异,通过比较共线性区域或重排区域的连续排列之间的 overlaps 和 gaps 来预测结构变异。
syri软件本身限制因素很多,主要是基因组必须是染色体版本、共线染色体需要都是正义链或都是负义链、染色体号需要一一对应等
syri软件安装也比较麻烦,需要是python3.5的环境,可以用conda安装需要支持的包
可以参考此博客,软件介绍比较详细:SyRI:一款从组装的基因组中检测结构变异的实用软件 - 简书 (jianshu.com)

 浙公网安备 33010602011771号
浙公网安备 33010602011771号