K近邻算法原理解析
K近邻算法基础
我们来介绍一下K近邻算法,这个算法应该说是机器学习中最简单的一个算法了,不过它虽然简单、但也有很多有点,比如:
思想极度简单使用的数学知识很少(近乎为0)对于一些特定的数据集有非常好的效果可以解释机器学习算法使用过程中的很多细节问题更完整地刻画机器学习应用的流程
那么什么是K近邻算法呢?我们来画一张图:
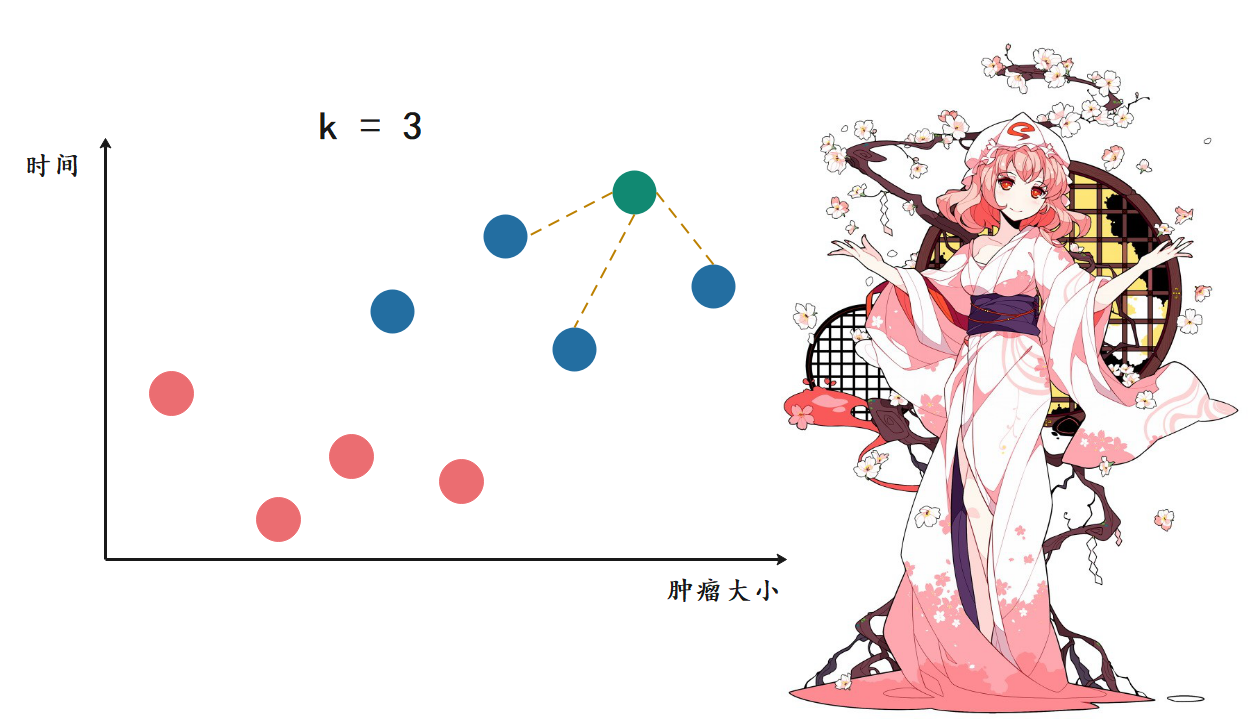
上图是以往病人体内的肿瘤状况,红色是良性肿瘤、蓝色是恶性肿瘤。显然这与发现时间的早晚以及肿瘤大小有密不可分的关系,那么当再来一个病人,我怎么根据时间的早晚以及肿瘤大小推断出这个新的病人体内的肿瘤(图中的绿色)是良性的还是恶性的呢?
K近邻的思想便可以在这里使用,我们可以根据距离(距离分为多种,我们后面会说,目前就使用图中所示的距离、也就是欧拉距离)选择离当前肿瘤最近的k个肿瘤,看看哪种肿瘤出现的次数最多。我们这里k取3,就看离自己最近的3个肿瘤,看看哪种出现的次数最多。显然它们都是蓝色,因此蓝色比红色等于3比0,蓝色出现的次数最多,因此通过K近邻算法我们就可以得出这个肿瘤是恶性的。
因此这便是K近邻,可以说其算法非常简单,就是寻找离当前样本最近的k个样本,看看这个k个样本中哪个类别出现的次数最多。所以K近邻思想,还是看样本的相似度,至于k到底为多少,显然没有一个统一的标准,这个是和你的数据集有很大关系的。当然在实际工作中,我们也不可能只取一个值,而是会将k取不同的值进行反复尝试。
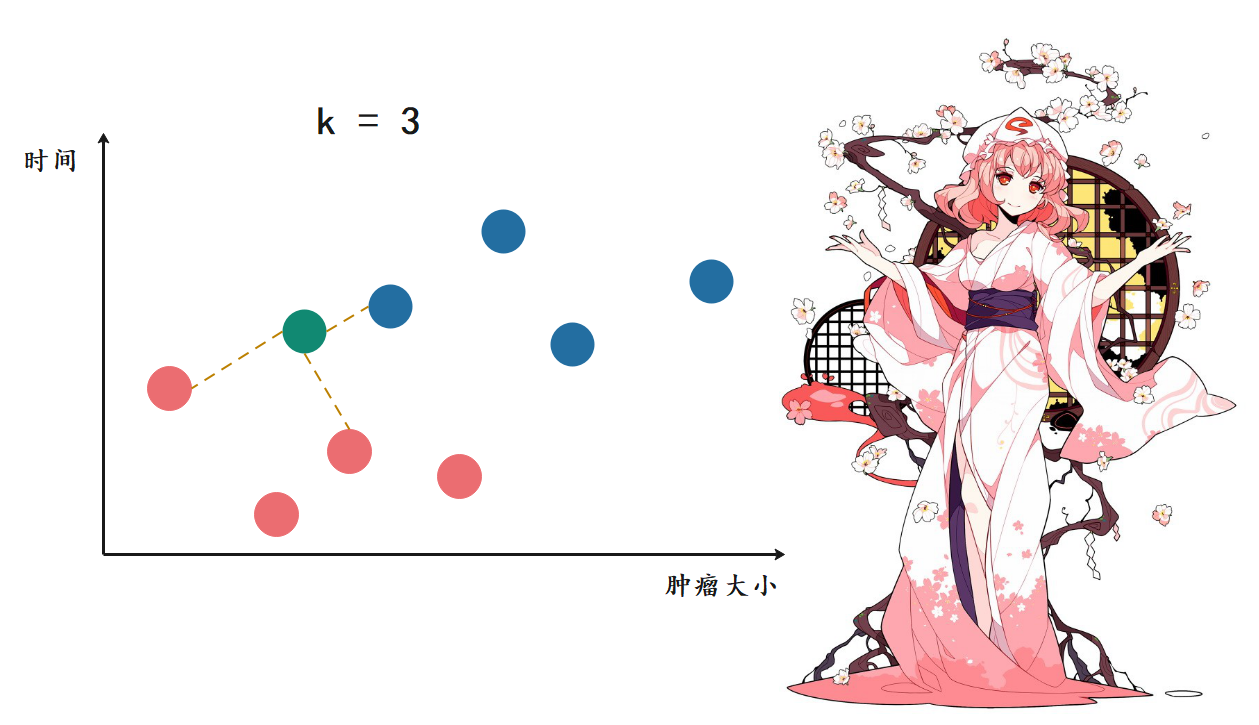
当再来一个肿瘤的时候,此时红色比蓝色等于2比1,红色胜出。那么K近邻就告诉我们,这个肿瘤很可能是一个良性肿瘤。
下面我们通过代码来实现一下K近邻,我们的代码都在jupyter notebook上运行。
注意:本文以及后续文章需要你熟悉Numpy。
import numpy as np
import plotly.graph_objs as go
# 可以看做肿瘤的发现大小和时间
data_X = [[3.39, 2.33],
[3.11, 1.78],
[1.34, 3.37],
[3.58, 4.68],
[2.28, 2.87],
[7.42, 4.70],
[5.75, 3.53],
[9.17, 2.51],
[7.79, 3.42],
[7.94, 0.79]]
# 可以看做是良性还是恶性
data_y = [0, 0, 0, 0, 0, 1, 1, 1, 1, 1]
# 转成numpy中的array
data_X, data_y = np.array(data_X), np.array(data_y)
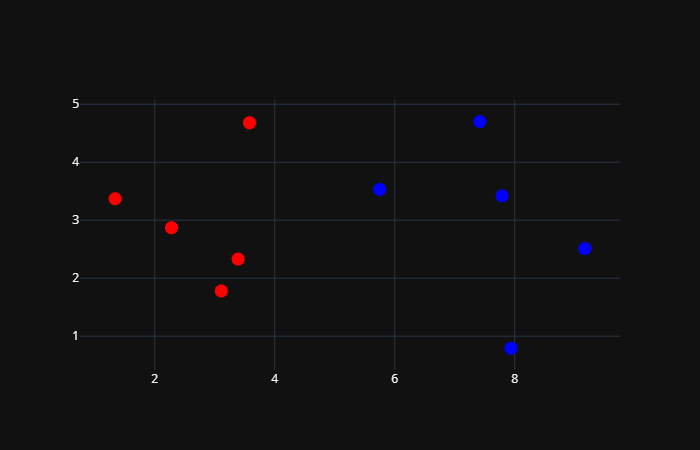
# 将良性肿瘤和恶性肿瘤对应的点都绘制在图中
# 良性肿瘤标记为红色,恶性肿瘤标记为蓝色
trace0 = go.Scatter(x=data_X[data_y == 0, 0],
y=data_X[data_y == 0, 1],
mode="markers",
marker={"size": 8, "color": "red"})
trace1 = go.Scatter(x=data_X[data_y == 1, 0],
y=data_X[data_y == 1, 1],
mode="markers",
marker={"size": 8, "color": "blue"})
fig = go.Figure(data=[trace0, trace1], layout={"template": "plotly_dark", "showlegend": False})
fig.show()
如果这个时候再来一个样本的话:
sample = np.array([[8.09, 3.37]])
trace0 = go.Scatter(x=data_X[data_y == 0, 0],
y=data_X[data_y == 0, 1],
mode="markers",
marker={"size": 13, "color": "red"})
trace1 = go.Scatter(x=data_X[data_y == 1, 0],
y=data_X[data_y == 1, 1],
mode="markers",
marker={"size": 13, "color": "blue"})
# 我们使用绿色绘制
trace2 = go.Scatter(x=sample[:, 0], y=sample[:, 1], mode="markers", marker={"size": 13, "color": "green"})
fig = go.Figure(data=[trace0, trace1, trace2], layout={"template": "plotly_dark", "showlegend": False})
fig.show()
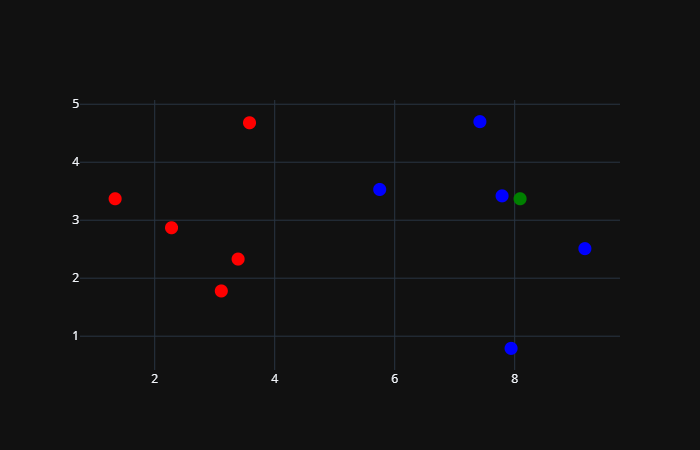
根据图像我们可以看出,这个新来的样本应该是属于红色这一类的。虽然从图像上我们大致能观察出来,但我们显然是要使用代码来实现的。至于实现方式也很简单,计算新的样本和已存在的所有样本之间的距离,然后选择最近(距离最短)的k个,找到出现次数最多的样本即可。
而距离我们选择欧拉距离,也就是两个点之间的直线距离,在坐标系上就是 "横坐标之差的平方 + 纵坐标之差的平方、再开根号" 。关于距离,我们后面还会详细说,除了欧拉距离还有其它的距离表示方式。
data_X = [[3.39, 2.33],
[3.11, 1.78],
[1.34, 3.37],
[3.58, 4.68],
[2.28, 2.87],
[7.42, 4.70],
[5.75, 3.53],
[9.17, 2.51],
[7.79, 3.42],
[7.94, 0.79]]
data_y = [0, 0, 0, 0, 0, 1, 1, 1, 1, 1]
data_X, data_y = np.array(data_X), np.array(data_y)
sample = np.array([[8.09, 3.37]]) # 新来的样本
distances = np.sqrt(np.sum((data_X - sample) ** 2, axis=1)) # 用于存放新来的样本和已存在的所有样本的距离
print(distances)
"""
[4.81368881
5.22766678
6.75
4.69640288
5.83147494
1.48922799
2.34546371
1.38057959
0.30413813
2.58435679]
"""
# 每一个样本 和 新样本之间的距离我们就计算出来了,然后我们排序选择最小的前三个
# 但是注意:此时的distances和data_y是一一对应的,所以我们不能对distances这个数组排序
# 如果对distances进行排序的话,那么它和data_y(样本标签)之间的对应的关系就被破坏了
# 因此我们需要按照值的大小来对索引排序
nearest = np.argsort(distances)
print(nearest) # [8 7 5 6 9 3 0 1 4 2]
# 表示离自己最近的是data_X中索引为8的样本,第二近的为索引为7的样本。
# 选择离自己最近的k个样本, 这里k取6, 然后找到它们的标签
topK_y = data_y[nearest[: 6]]
from collections import Counter
# 进行投票,选择出现次数最多的标签
votes = Counter(topK_y).most_common(1)[0]
print(votes) # (1, 5)
print(f"标签{votes[0]}出现次数最多,出现{votes[1]}次") # 标签1出现次数最多,出现5次
以上便是我们简单地使用K近邻算法对样本数据进行的一个简单预测,你也可以自己尝试一下。当然了,我们还可以对其进行一个封装。
def knn_classify(k: int,
data_X: np.ndarray,
data_y: np.ndarray,
sample: np.ndarray) -> str:
"""
:param k: 选择多少个样本进行投票
:param data_X: 训练样本
:param data_y: 标签
:param sample: 预测样本
:return:
"""
assert 1 <= k <= X_train.shape[0], "K必须大于等于1,并且小于等于训练样本的总数量"
assert X_train.shape[0] == y_train.shape[0], "训练样本的数量必须和标签数量保持一致"
assert x.shape[0] == X_train.shape[1], "预测样本的特征数必须和训练样本的特征数保持一致"
distances = np.sqrt(np.sum((data_X - sample) ** 2, axis=1))
nearest = np.argsort(distances)
topK_y = y_train[nearest[: k]]
votes = Counter(topK_y).most_common(1)[0]
return f"该样本的特征为{votes[0]},在{k}个样本中出现了{votes[1]}次"
data_X = [[3.39, 2.33],
[3.11, 1.78],
[1.34, 3.37],
[3.58, 4.68],
[2.28, 2.87],
[7.42, 4.70],
[5.75, 3.53],
[9.17, 2.51],
[7.79, 3.42],
[7.94, 0.79]]
data_y = [0, 0, 0, 0, 0, 1, 1, 1, 1, 1]
data_X = np.array(data_X)
data_y = np.array(data_y)
sample = np.array([8.09, 3.37]) # 新来的样本
print(knn_classify(3, data_X, data_y, sample)) # 该样本的特征为1,在3个样本中出现了3次
到目前为止,我们便通过代码简单地实现了一下K近邻算法。
什么机器学习
我们说k近邻算法是一种机器学习算法,那么机器学习是什么呢?机器学习就是我们输入大量的学习资料,然后通过机器学习算法(k近邻只是其中一种)训练出一个模型,当下一个样例到来的时候,我们预测出结果。
我们输入一定数量的样本(训练数据集),每个样本里面包含了X_train(样本特征)和y_train(样本标签),通过机器学习算法,对样本进行训练得到模型(特征和标签之间的对应关系),这一过程我们称之为fit(拟合);然后输入新的样本根据上一步得到的模型来预测出结果,这一过程我们称之为predict(预测)。
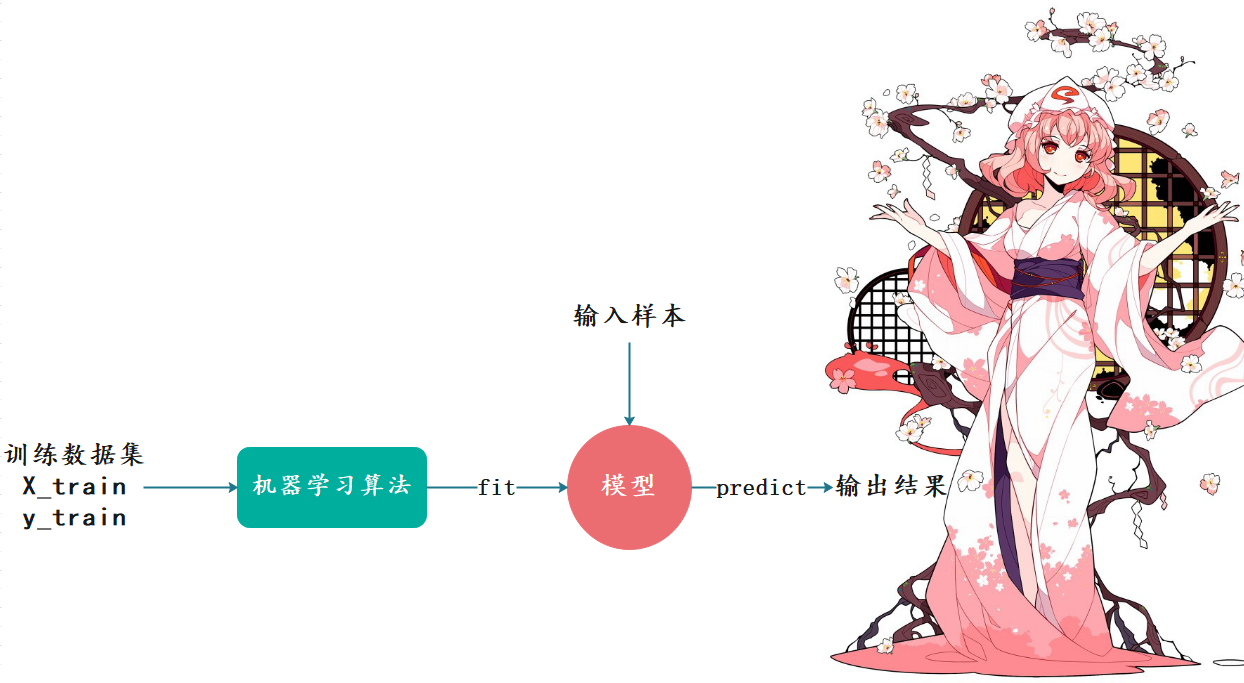
估计有人注意到了,我们在k近邻算法中,并没有看到训练模型这一步啊。可以这么说,k近邻是一个不需要训练过程的算法。K近邻算法是非常特殊的,可以认为是一个没有模型的算法。不过为了和其他算法统一,可以认为训练数据集本身就是模型。
因此我们还是为K近邻找到了一个fit的过程,这在sklearn中也是这么设计的。sklearn里面封装了大量的算法,它们的设计理念都是一样的,都是先fit训练模型,然后predict进行预测。
sklearn中的K近邻算法
我们看看如何在sklearn中使用K近邻算法
import numpy as np
from sklearn.neighbors import KNeighborsClassifier
# sklearn里面所有的算法都是以面向对象的形式封装的
knn = KNeighborsClassifier(n_neighbors=3) # 这里的n_neighbors就是k近邻中的那个k
raw_data_X = [[3.39, 2.33],
[3.11, 1.78],
[1.34, 3.37],
[3.58, 4.68],
[2.28, 2.87],
[7.42, 4.70],
[5.75, 3.53],
[9.17, 2.51],
[7.79, 3.42],
[7.94, 0.79]]
raw_data_y = [0, 0, 0, 0, 0, 1, 1, 1, 1, 1]
X_train = np.array(raw_data_X) # 样本特征
y_train = np.array(raw_data_y) # 样本标签
# 传入数据集进行fit,得到模型
knn.fit(X_train, y_train)
# 使用模型对新来的样本进行predict
# 注意:在sklearn中,predict必须要传递一个二维数组,哪怕只有一个元素,也要写成二维数组的模式
y_predict = knn.predict(np.array([[8.09, 3.37],
[3.09, 7.37]]))
# 由此我们得到这两个样本的标签分别是1和0
print(y_predict) # [1, 0]
总结一下就是:
每个算法在sklearn中就是一个类,通过给类指定参数(不指定会用默认的)得到一个实例调用实例的fit方法,传入训练数据集的特征和标签,来训练出一个模型。并且这个方法会返回实例本身,当然我们也并不需要再单独地使用变量接收fit完毕之后,直接调用predict,得到预测的结果。并且注意的是,在sklearn中,要传入二维数组,哪怕只预测一个样本也要以二维数组的形式传递。比如:不要传递np.array([8.09, 3.37]),而应该传递np.array([[8.09, 3.37]])保存得到的预测值
在sklearn中所有的算法都保持这样的高度一致性,都是得到实例,再fit,然后predict。
封装自己的K近邻
显然以这种类的模式,是更加友好的。那么我们也可以将我们之间写的knn算法,封装成sklearn中的模式,注意:K近邻算法也被称为Knn。
from collections import Counter
import numpy as np
class KnnClassifier:
def __init__(self, k):
assert k >= 1, "K必须合法"
self.k = k
# 用户肯定会传入样本集,因此提前写好。使用_开头表示这些变量不建议从外部访问。
self._X_train = None
self._y_train = None
def fit(self, X_train, y_train):
assert X_train.shape[0] == y_train.shape[0], "训练样本的数量必须和标签数量保持一致"
assert self.k <= X_train.shape[0], "K必须小于等于训练样本的总数量"
# 因为K近邻算法是一个不需要训练模型的算法,这里我们没有训练这一步
self._X_train = X_train
self._y_train = y_train
return self
# 这里完全可以不需要return self,但是为了和sklearn中的格式保持一致,我们还是加上这句
# 为什么这么做呢?其实如果我们严格按照sklearn的标准来写的话,那么我们自己写的算法是可以和sklearn中的其他算法无缝对接的
def predict(self, X_predict):
assert self._X_train is not None and self._y_train is not None, "在predict之间必须先进行fit"
if not isinstance(X_predict, np.ndarray):
raise TypeError("传入的数据集必须是np.ndarray类型")
predict = []
# 因为可能来的不止一个样本
for x in X_predict:
distances = np.sqrt(np.sum((self._X_train - x) ** 2, axis=1))
nearest = np.argsort(distances)
votes = self._y_train[nearest[: self.k]]
predict.append(Counter(votes).most_common(1)[0][0])
# 遵循sklearn的标准,以np.ndarry的形式返回
return np.array(predict)
def __repr__(self):
return f"KNN(k={self.k})"
def __str__(self):
return f"KNN(k={self.k})"
raw_data_X = [[3.39, 2.33],
[3.11, 1.78],
[1.34, 3.37],
[3.58, 4.68],
[2.28, 2.87],
[7.42, 4.70],
[5.75, 3.53],
[9.17, 2.51],
[7.79, 3.42],
[7.94, 0.79]]
raw_date_y = [0, 0, 0, 0, 0, 1, 1, 1, 1, 1]
X_train = np.array(raw_data_X)
y_train = np.array(raw_date_y)
x = np.array([[8.09, 3.37],
[5.75, 3.53]]) # 新来的点
knn = KnnClassifier(k=3)
knn.fit(X_train, y_train)
print(knn)
y_predict = knn.predict(x)
print(y_predict)
运行结果:
KNN(k=3)
[1 1]
怎么样,是不是很简单呢?我们以上便实现了一个自己的Knn。
训练数据集、测试数据集
我们目前训练模型是将所有的数据集都拿来训练模型了,但是问题来了,如果模型很差怎么办?首先我们没有机会去调整,因为我们的模型是要在真实的环境中使用的。因此这种方式我们就无法得知模型的好坏,在真实的环境中使用就只能听天由命了。其实说这些,意思就是将全部的数据集都用来寻来模型是不合适的,因此我们可以将数据集进行分割。比如百分之80的数据集用来训练,百分之20的数据集用于测试。
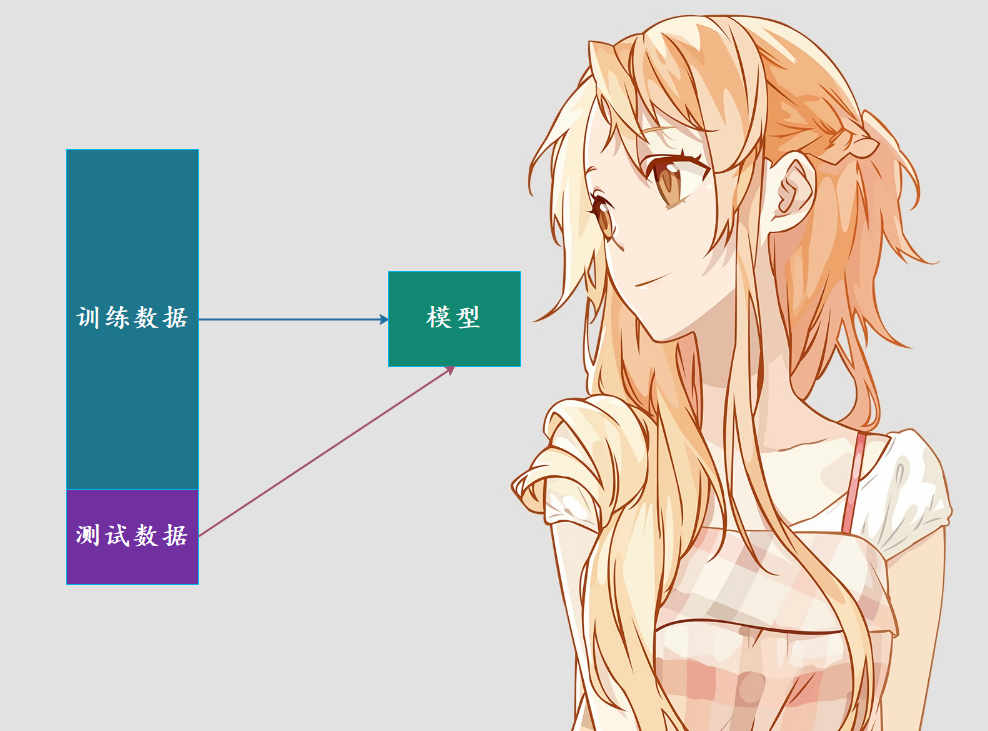
通过测试数据直接判断模型好坏,在模型进入真实环境之前改进模型。
如果对测试数据集预测的不够好的话,说明我们的算法还需要改进,这样就可以在进入真实环境之前改进模型。而将整个数据集分割成训练数据集合测试数据集这一过程我们称之为train_test_split。而sklearn也提供了这样一个函数,不过我们也可以自己手动模拟一下。
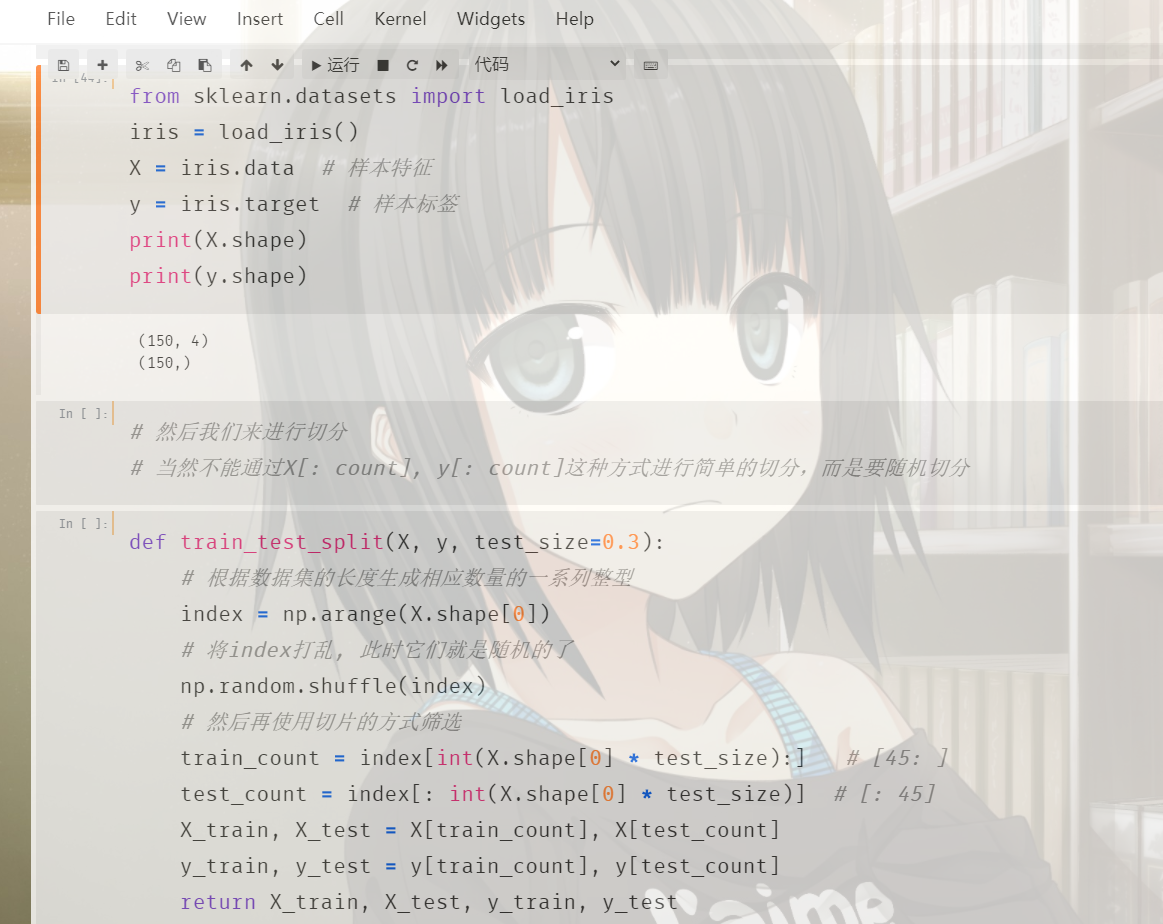
将我们之前自己写的knn算法和切割数据集的代码组合起来,一部分用于训练,一部分用于预测。
from collections import Counter
import numpy as np
from sklearn.datasets import load_iris
class KnnClassifier:
def __init__(self, k):
assert k >= 1, "K必须合法"
self.k = k
self._X_train = None
self._y_train = None
def fit(self, X_train, y_train):
assert X_train.shape[0] == y_train.shape[0], "训练样本的数量必须和标签数量保持一致"
assert self.k <= X_train.shape[0], "K必须小于等于训练样本的总数量"
self._X_train = X_train
self._y_train = y_train
return self
def predict(self, X_predict):
assert self._X_train is not None and self._y_train is not None, "在predict之间必须先进行fit"
if not isinstance(X_predict, np.ndarray):
raise TypeError("传入的数据集必须是np.ndarray类型")
predict = []
for x in X_predict:
distances = np.sqrt(np.sum((self._X_train - x) ** 2, axis=1))
nearest = np.argsort(distances)
votes = self._y_train[nearest[: self.k]]
predict.append(Counter(votes).most_common(1)[0][0])
return np.array(predict)
def __repr__(self):
return f"KNN(k={self.k})"
def __str__(self):
return f"KNN(k={self.k})"
def train_test_split(X, y, test_size=0.3):
index = np.arange(X.shape[0])
np.random.shuffle(index)
train_count = index[int(X.shape[0] * test_size):] # [45: ]
test_count = index[: int(X.shape[0] * test_size)] # [: 45]
X_train, X_test = X[train_count], X[test_count]
y_train, y_test = y[train_count], y[test_count]
return X_train, X_test, y_train, y_test
iris = load_iris()
X = iris.data # 样本特征
y = iris.target # 样本标签
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3)
knn.fit(X_train, y_train)
# 预测
y_predict = knn.predict(X_test)
# 将对测试集进行预测的结果和已知的结果进行比对
print(f"测试样本总数:{len(X_test)}, "
f"预测正确的数量:{np.sum(y_predict == y_test)}, "
f"预测准确率:{np.sum(y_predict == y_test) / len(X_test)}")
运行结果:
测试样本总数:45, 预测正确的数量:44, 预测准确率:0.9777777777777777
最后来看看sklearn中提供的train_test_split。
from sklearn.model_selection import train_test_split
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3)
用法和我们自定义的一样,而且sklearn中的train_test_split可以接收更多的参数,比如我们还可以设置一个random_state,表示随机种子。
因此以上就是训练集和测试集的内容,训练集进行训练,测试集进行测试。如果在测试集上表现完美的话,我们才会投入生产环境使用,如果表现不完美,那么我们可能就要调整我们的算法,或者调整我们的参数了
分类准确度
评价一个算法的好坏,我们可以使用分类准确度来实现。如果预测正确的样本个数越多,那么说明我们的算法越好。所以我们可以我们自己的Knn算法中定义一个score方法,根据预测的准确度来给模型打一个分。
from collections import Counter
import numpy as np
from sklearn.datasets import load_digits
from sklearn.model_selection import train_test_split
class KnnClassifier:
def __init__(self, k):
assert k >= 1, "K必须合法"
self.k = k
self._X_train = None
self._y_train = None
def fit(self, X_train, y_train):
assert X_train.shape[0] == y_train.shape[0], "训练样本的数量必须和标签数量保持一致"
assert self.k <= X_train.shape[0], "K必须小于等于训练样本的总数量"
self._X_train = X_train
self._y_train = y_train
return self
def predict(self, X_predict):
assert self._X_train is not None and self._y_train is not None, "在predict之间必须先进行fit"
if not isinstance(X_predict, np.ndarray):
raise TypeError("传入的数据集必须是np.ndarray类型")
predict = []
for x in X_predict:
distances = np.sqrt(np.sum((self._X_train - x) ** 2, axis=1))
nearest = np.argsort(distances)
votes = self._y_train[nearest[: self.k]]
predict.append(Counter(votes).most_common(1)[0][0])
return np.array(predict)
def score(self, X_test, y_test):
y_predict = self.predict(X_test)
# 预测正确的个数除以总个数
return np.sum(y_test == y_predict) / len(y_test)
"""
还可以这么做
from sklearn.metrics import accuracy_score
return accuracy_score(y_test, y_predict)
"""
def __repr__(self):
return f"KNN(k={self.k})"
def __str__(self):
return f"KNN(k={self.k})"
# 使用sklearn中的手写字体
digits = load_digits()
X = digits.data
y = digits.target
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3)
knn = KnnClassifier(k=3)
knn.fit(X_train, y_train)
print(knn.score(X_test, y_test)) # 0.9907407407407407
我们通过封装一个score方法,直接对模型打分。当然你可以调用predict获取预测值,然后将预测值和真实值进行比对,手动计算分数;但如果只想知道模型的好坏,那么可以调用score方法,传入测试数据的X_test和y_test,然后score方法内部会自动调用predict,根据X_test预测出y_predict,然后通过y_predict和y_test计算出分数。
超参数
对于已经实现好的算法,我们一般不会重复造轮子,当然一些特定场景除外。不过我们在使用sklearn的K近邻算法(KNeighborsClassifier)的时候,貌似只传递了一个参数:n_neighbors,也就是K近邻中的k。实际上这个n_neighbors参数也是可以不传的,因为会有一个默认值,默认值是5。那么除了n_neighbors之外还有没有其它的参数呢?答案是肯定有,而这些参数就是超参数。超参数,指的是那些在fit之前,就需要指定的参数。
说起超参数就不得不提模型参数,这两者区别是什么呢?
超参数:在算法运行前(或者说在fit之前)需要决定的参数模型参数:算法过程中学习的参数
Knn算法比较特殊,它没有模型参数,因为数据集本身就可以看做成一个模型。但是后续的算法中,比如线性回归算法,都会包含大量的模型参数。Knn算法中的k是典型的超参数
那么如何寻找更好的超参数呢?
在机器学习中,我们经常会听到一个词叫"调参",这个调参调的就是超参数,因为这是在算法运行前就需要指定好的参数。而寻找好的超参数一般有三种办法:
领域知识:这个是根据所处的领域来决定的,比如文本处理,视觉领域等等,不同的领域会有不同的超参数经验数值:根据以往的经验,比如sklearn的knn算法中,默认的k就是5,这个是根据经验决定的。另外sklearn的每个算法中都有大量的超参数,并且大部分都有默认值,这些默认值是sklearn根据以往的经验选择的最佳的默认值。实验搜索:如果无法决定的话,那么就只能通过实验来进行搜索了。我们对超参数取不同的值进行测试、对比,寻找一个最好的超参数。
我们来暴力搜索一下吧:
from sklearn.datasets import load_digits
from sklearn.model_selection import train_test_split
from sklearn.neighbors import KNeighborsClassifier
digits = load_digits()
X = digits.data
y = digits.target
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3, random_state=666)
def search_best_k():
best_score = 0.0
best_k = -1
for k in range(1, 11):
# k分别从1取到10
knn_clf = KNeighborsClassifier(n_neighbors=k)
knn_clf.fit(X_train, y_train) # fit
score = knn_clf.score(X_test, y_test) # 得到score
if score > best_score:
best_score = score
best_k = k
return f"the best score is {best_score}, k is {best_k}"
print(search_best_k()) # the best score is 0.9888888888888889, k is 6
以上便是调参的过程,通过对不同的超参数取值,确定一个最好的超参数。当然我的机器以及版本得出来的是6,可能不同的人计算之后,得到的结果会有所差异。
但是当我们得到k是10的话,这就意味着此时的k值是处于边界,那么我们不确定边界之外的值对算法的影响如何,可能10以后的效果会更好。因此我们还需要对10以外的值进行判断,在参数值取到我们给定范围的边界时,我们需要稍微地再拓展一下,扩大搜索范围,对边界之外的值进行检测、判断,看看能否得到更好的超参数。
K近邻的其他超参数
实际上,knn不止一个超参数,还有一个很重要的超参数,那就是距离。还以肿瘤为例:
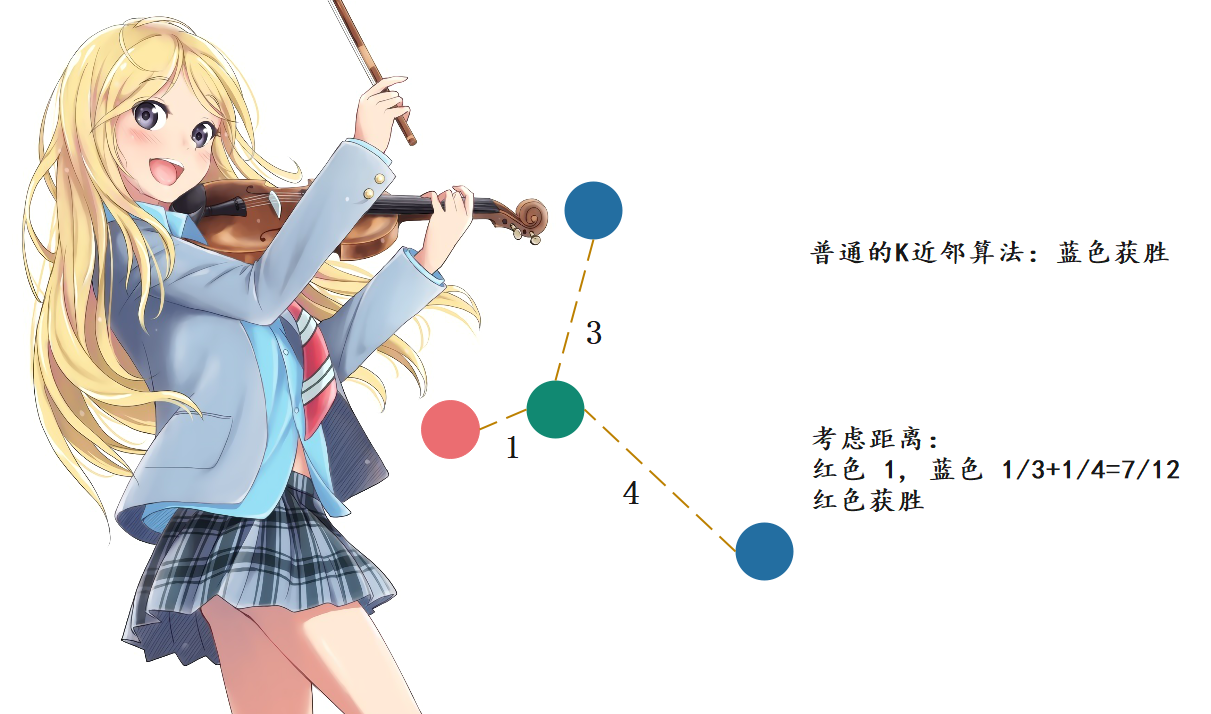
按照我们之前讲的,在k取3的时候,蓝色有两个,红色有1个,那么蓝色比红色是2比1,所以蓝色获胜。但是我们忽略了一个问题,那就是距离,这个绿色是不是离红色更近一些呢?一般肿瘤发现时间和大小比较接近的话,那么肿瘤的性质也类似。比如红色,绿色和红色显然离得近一些,那么理论上结果也更相似一些,但是我们选择了3个,还有两个是蓝色的,但是两者都离绿色比较远。因此如果还用个数投票的话,那么显然是不公平的,因此距离近的,那么相应的权重是不是也要更大一些呢?因此我们可以采用距离的倒数进行计算,这样做的话会发现是红色胜出。
另外采用距离的倒数,还可以有一个好处,那就是解决平票的问题。假设绿色周围都是不同的颜色,那么我们依旧可以根据距离得出结果;如果是根据数量,那么显然每个样本都是1,此时就平票了。
那么下面我们看一下sklearn中的KNeighborsClassifier都有哪些参数。
class KNeighborsClassifier(NeighborsBase, KNeighborsMixin,
SupervisedIntegerMixin, ClassifierMixin):
def __init__(self, n_neighbors=5,
weights='uniform', algorithm='auto', leaf_size=30,
p=2, metric='minkowski', metric_params=None, n_jobs=None,
**kwargs):
n_neighbors=5: knn当中的k,指定以几个最邻近的样本具有投票权weights:每个拥有投票权的样本是按照什么比重投票。默认是uniform表示等比重,也就是不考虑距离,每一个成员的权重都是一样的。还可以指定为distance,就是我们之前说的按照距离的反比进行投票algorithm="auto":内部采用什么样的算法实现,有以下几种方法,"ball_tree":球树,"kd_tree":kd树,"brute":暴力搜索。"auto"表示自动根据数据类型和结构选择合适的算法。一般来说,低维数据用kd_tree,高维数据用ball_treeleaf_size=30:ball_tree或者kd_tree的叶子节点规模matric="minkowski":怎样度量距离,默认是闵可夫斯基距离p=2:闵式距离中的距离参数, 后面会说metric_params=None:距离度量函数的额外关键字参数,一般默认为None,不用管n_jobs=None: 并行的任务数,默认使用1个核心,指定为-1则使用所有核心
一般来说n_neighbors、weights、p这三个参数对于调参来说是最常用的。
下面我们就来考虑n_neighbors和weights这个两个参数。
from sklearn.datasets import load_digits
from sklearn.model_selection import train_test_split
from sklearn.neighbors import KNeighborsClassifier
digits = load_digits()
X = digits.data
y = digits.target
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3, random_state=666)
def search_best_k():
best_weight = ""
best_score = 0.0
best_k = -1
for weight in ["uniform", "distance"]:
for k in range(1, 11):
knn_clf = KNeighborsClassifier(n_neighbors=k, weights=weight)
knn_clf.fit(X_train, y_train)
score = knn_clf.score(X_test, y_test)
if score > best_score:
best_score = score
best_k = k
best_weight = weight
return f"the best score is {best_score}, k is {best_k}, weight is {best_weight}"
print(search_best_k()) # the best score is 0.9888888888888889, k is 6, weight is uniform
将距离考虑进去之后,得到的结果。
更多关于距离的定义
我们之前讨论距离,显然都是欧拉距离。说白了就是两点确认一条直线,这条直线的长度。那么下面我们介绍更多的距离,比如sklearn的knn的参数中的闵可夫斯基距离(闵式距离)。
1. 欧拉距离:
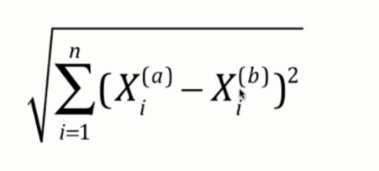
这个无需介绍,就是两个点连接的直线的距离。
2. 曼哈顿距离:

曼哈顿距离,指的是各个维度上的距离之和,也就是图中蓝色的距离,或者说是红色的距离、黄色的距离,三者都是一样的,各个维度上的距离之和就是曼哈顿距离。那么图中的绿色的那条线显然就是欧拉距离。
3. 闵可夫斯基距离:
首先曼哈顿距离是每个维度的距离之和;欧拉距离是每个维度的距离的平方之和再开平方;闵可夫斯基距离则是每个维度的距离的p次方之和再开p次方。
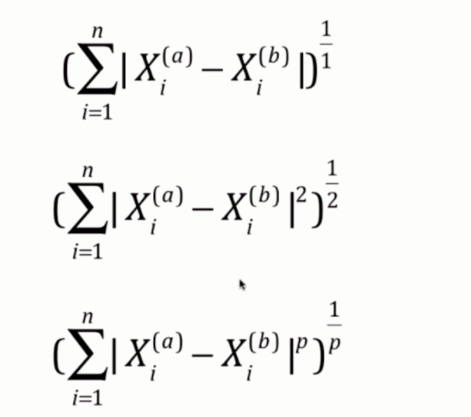
因此曼哈顿距离和欧拉距离可以看成是闵可夫斯基距离的p分别取1和2所对应的情况。在sklearn的KNeighborsClassifier中,里面就有一个参数p,这个p指的就是图中闵式距离里面的p。而p默认取2,那么显然默认是欧拉距离。
使用numpy计算这几个距离:
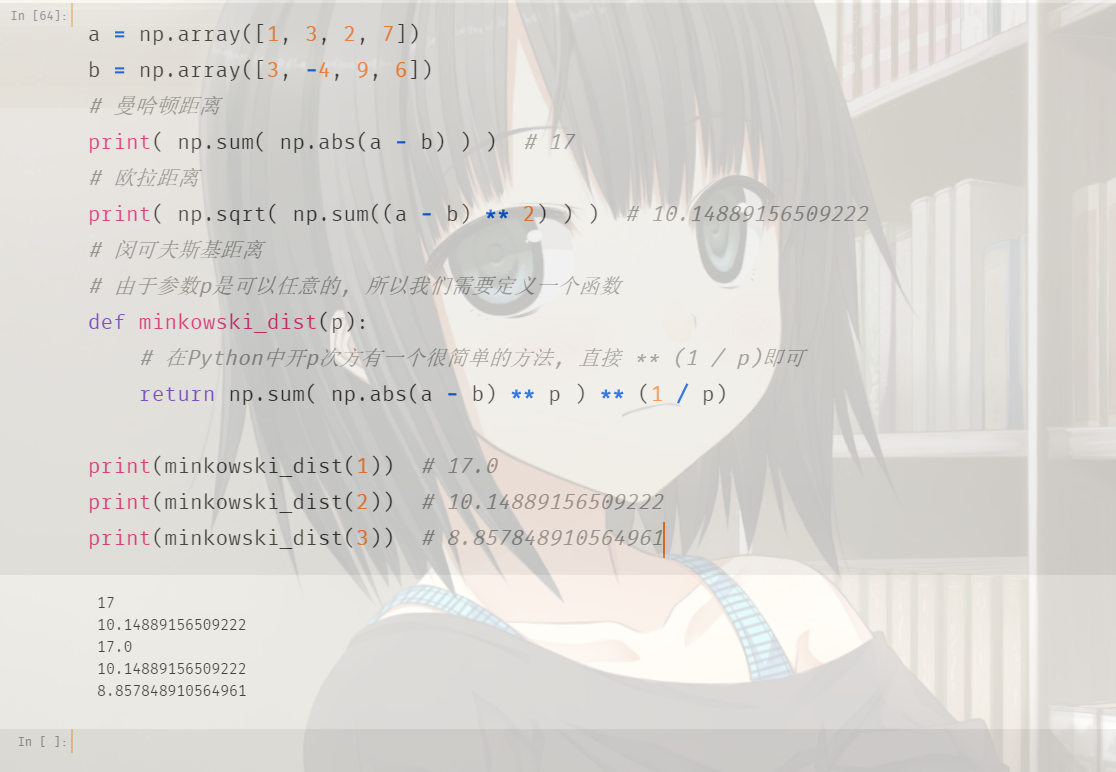
比较简单,不多说了。
搜索最好的参数p
当我们将weights设置为"distance"的时候,显然是需要将p也考虑在内的。
from sklearn.datasets import load_digits
from sklearn.model_selection import train_test_split
from sklearn.neighbors import KNeighborsClassifier
digits = load_digits()
X = digits.data
y = digits.target
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3, random_state=666)
def search_best_k():
best_p = -1
best_score = 0.0
best_k = -1
for k in range(1, 11):
# 既然指定了p,显然是考虑距离的,那么weights就必须直接指定为distance,不然p就没有意义了
for p in range(1, 6):
knn_clf = KNeighborsClassifier(n_neighbors=k, weights="distance", p=p)
knn_clf.fit(X_train, y_train)
score = knn_clf.score(X_test, y_test)
if score > best_score:
best_score = score
best_k = k
best_p = p
return f"the best score is {best_score}, k is {best_k}, p is {best_p}"
print(search_best_k()) # the best score is 0.9888888888888889, k is 5, p is 2
我们看到,找到的k是5、p是2,这和默认参数是一致的,因此sklearn参数的默认值设置的还是比较好的,当然sklearn中参数的默认值都是基于以往的规律总结得到的。
不过对于像p这种参数,说实话一般不常用。比如sklearn中其他的算法,都有很多超参数。但有的参数我们一般很少会用,而且这些参数都会有一些默认值,一般都是sklearn根据经验给你设置的最好或者比较好的默认值,这些参数不需要改动。比如knn中的algorithm和leaf_size,这些真的很少会使用,直接使用默认的就行。但是像n_neighbors之类的,放在参数最前面的位置,肯定是重要的。我们需要了解原理、并且在搜索中也是会经常使用的。
这个搜索过程就叫做网格搜索,比如图中的k和p,k、p取不同的值,这些值按照左右和上下的方向连起来就形成了一个网格,网格上的每个点所代表的就是每一个k和p。
网格搜索与K近邻算法中更多超参数
事实上,超参数之间也是有依赖的。比如在knn中,我们如果指定p的话,那么就必须要将weights指定为"distance",如果不指定,那么p就没有意义了。因此这种情况比较麻烦,怎么才能将我们所有的参数都列出来,只运行一次就得到最好的超参数呢?显然自己实现是完全可以的,就再写一层for循环嘛,但是sklearn为我们这种网格搜索,专门封装了一个函数。使用这个函数,就能更加方便地找到最好的超参数。
from sklearn.datasets import load_digits
from sklearn.model_selection import train_test_split
from sklearn.neighbors import KNeighborsClassifier
from sklearn.model_selection import GridSearchCV
digits = load_digits()
X = digits.data
y = digits.target
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3, random_state=666)
# 定义网格搜索的参数
# 参数以字典的形式传入,由于会有多个值,因此value都要是以列表(可迭代)的形式,哪怕只有一个参数,也要以列表的形式
# 之前说了有些参数是依赖于其他参数的,比如这里的p,必须是在weights="distance"的时候才会起作用。但是weights又不仅仅只能取distance
# 那么就可以分类,将多个字典放在一个列表里面。即便只有一个字典,也要放在列表里面。
# 总结一下就是:[{}, {}, {}]
param_grid = [
{
"weights": ["uniform"],
"n_neighbors": list(range(1, 11))
},
{
"weights": ["distance"],
"n_neighbors": list(range(1, 11)),
"p": list(range(1, 6))
}
]
# 创建一个实例,不需要传入任何参数
knn = KNeighborsClassifier()
# 会发现这个类后面有一个CV,这个CV暂时不用管,后面介绍。
# 创建一个实例,里面传入我们创建的estimator和param_grid即可。
# estimator是啥?我们之前说过,每一个算法在sklearn中都被封装成了一个类,根据这个类创建的实例就叫做estimator(分类器)。
grid_search = GridSearchCV(knn, param_grid, n_jobs=-1) # n_jobs=-1表示使用所有的核进行搜索
# 让grid_search来fit我们的训练集,会用不同的超参数来进行fit,这些超参数就是我们在param_grid当中指定的超参数
grid_search.fit(X_train, y_train)
# 打印最佳的分数
print(grid_search.best_score_)
# 打印最佳的参数
print(grid_search.best_params_)
运行结果:
0.9920445203313729
{'n_neighbors': 1, 'weights': 'uniform'}
以上我们便找到了最佳的分数和最佳的参数,在weights为"distance"的时候,我们知道最好的超参数是n_neightbors为5、p为2,但是显然将weights设置为默认的"uniform"会更合适一些,此时n_neightbors为1。我们直接将grid_search.best_params_这个字典打散传入到KNeighborsClassifier中即可得到使用最好的超参数创建的estimator。当然sklearn也帮我们想到了这一点,提前已经帮我们做好了。
# 打印最佳的分类器
print(grid_search.best_estimator_)
"""
KNeighborsClassifier(algorithm='auto', leaf_size=30, metric='minkowski',
metric_params=None, n_jobs=None, n_neighbors=1, p=2,
weights='uniform')
"""
# 等价于:KNeighborsClassifier(**grid_search.best_params_)
因此这便是网格搜索,说白了就是暴力尝试,如果想查看更详细的信息可以在GridSearchCV中加上一个verbose参数。这个参数表示输出详细信息,接收一个数字,值越大,输出信息越详细,一般传入2就行。有兴趣可以自己尝试一下,看看都输出了哪些信息。
另外,我们看到像best_score_、best_params_、best_estimator_这些参数的结尾都加上了一个下划线,这是sklearn中的一种命名方式。结尾带有下划线的变量,表示它不是由用户传递的,而是根据其它参数在fit的时候生成的,并且还可以提供给外界进行访问,这样的变量会在结尾处加上一个下划线。
数据归一化
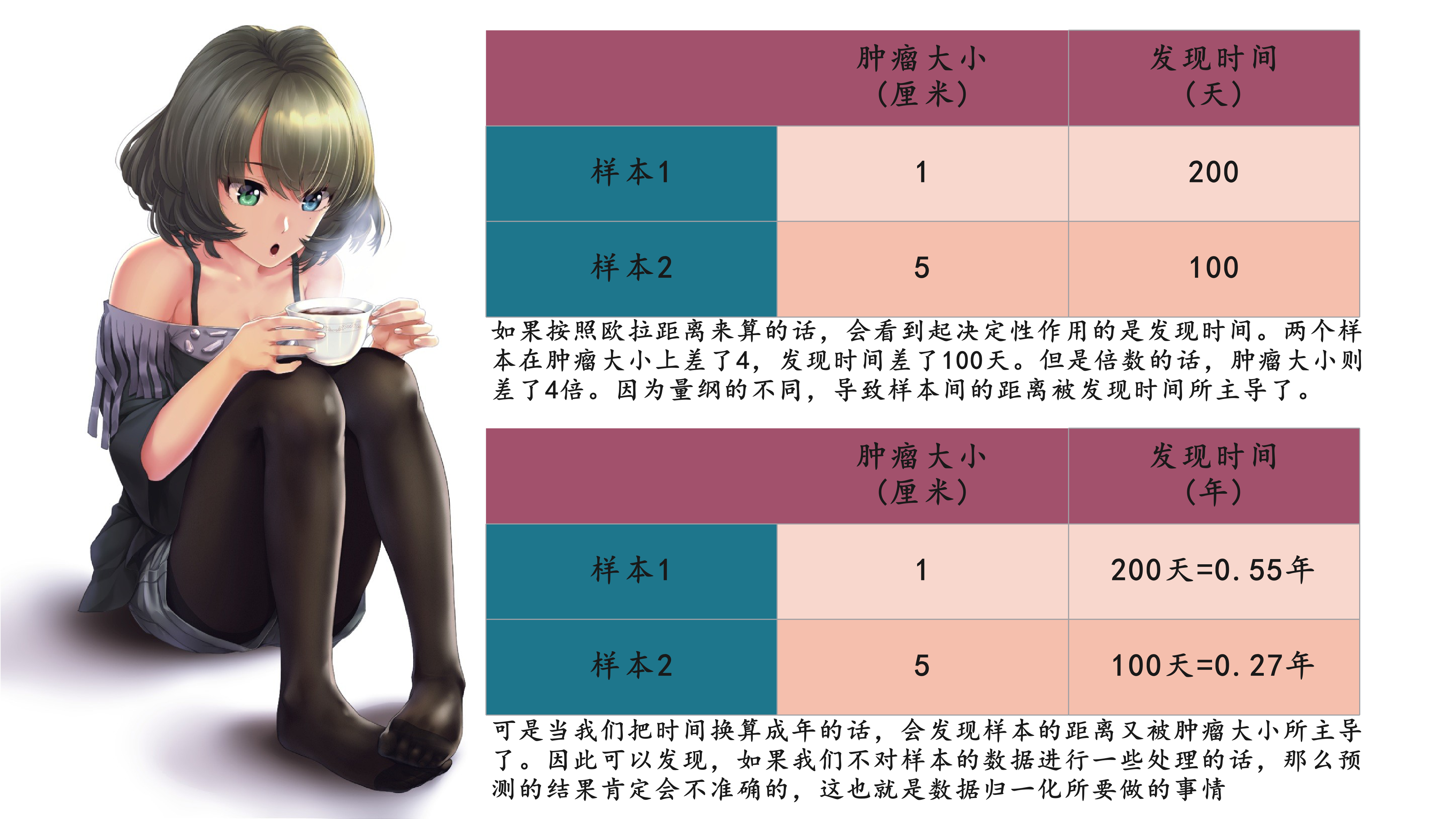
有些时候,不同维度的数据的量纲是不一样的,就拿肿瘤的例子来举例。
那么数据归一化都有哪些手段呢?
最值归一化:将所有的数据都映射到0 ~ 1之间。
\(x_{scale} = \frac{x - x_{min}}{x_{max} - x_{min}}\)
但是这样有一个问题,就是适用于有明显边界的情况,比如学生的考试成绩,一般是0到100分;再比如图像的像素点,一般是0到255。这些情况是适合最值归一化的。而对于那些没有明显边界的情况的话,则不适合了,比如收入。大部分人收入假设是一个月一万元,但是一个哥们比较niubility,他月薪十万。因此边界过大,那么大部分人就都被映射到了0.1左右,显然这种情况是不适合最值归一化的。
import numpy as np
x = np.random.randint(0, 100, size=100)
print(x)
"""
[73 31 88 99 70 42 30 57 5 30 60 58 6 72 18 66 36 7 93 66 30 36 4 0
68 8 70 85 23 11 68 70 58 95 73 38 38 35 66 13 4 77 23 42 18 31 97 82
63 53 42 41 97 7 38 41 28 9 97 42 71 30 50 81 80 91 3 75 3 33 42 14
19 68 59 37 36 51 92 30 58 57 87 98 58 14 23 0 33 63 15 90 25 16 78 59
51 60 24 30]
"""
x_max = np.max(x)
x_min = np.min(x)
x = (x - x_min) / (x_max - x_min)
print(x)
"""
[0.73737374 0.31313131 0.88888889 1. 0.70707071 0.42424242
0.3030303 0.57575758 0.05050505 0.3030303 0.60606061 0.58585859
0.06060606 0.72727273 0.18181818 0.66666667 0.36363636 0.07070707
0.93939394 0.66666667 0.3030303 0.36363636 0.04040404 0.
0.68686869 0.08080808 0.70707071 0.85858586 0.23232323 0.11111111
0.68686869 0.70707071 0.58585859 0.95959596 0.73737374 0.38383838
0.38383838 0.35353535 0.66666667 0.13131313 0.04040404 0.77777778
0.23232323 0.42424242 0.18181818 0.31313131 0.97979798 0.82828283
0.63636364 0.53535354 0.42424242 0.41414141 0.97979798 0.07070707
0.38383838 0.41414141 0.28282828 0.09090909 0.97979798 0.42424242
0.71717172 0.3030303 0.50505051 0.81818182 0.80808081 0.91919192
0.03030303 0.75757576 0.03030303 0.33333333 0.42424242 0.14141414
0.19191919 0.68686869 0.5959596 0.37373737 0.36363636 0.51515152
0.92929293 0.3030303 0.58585859 0.57575758 0.87878788 0.98989899
0.58585859 0.14141414 0.23232323 0. 0.33333333 0.63636364
0.15151515 0.90909091 0.25252525 0.16161616 0.78787879 0.5959596
0.51515152 0.60606061 0.24242424 0.3030303 ]
"""
同理对于多维数组也是可以的。
import numpy as np
x = np.random.randint(0, 100, size=(10, 2))
print(x)
"""
[[60 38]
[14 85]
[17 56]
[ 4 7]
[21 65]
[69 25]
[48 85]
[98 0]
[24 8]
[43 78]]
"""
x_max = np.max(x, axis=0)
x_min = np.min(x, axis=0)
x = (x - x_min) / (x_max - x_min)
print(x)
"""
[[0.59574468 0.44705882]
[0.10638298 1. ]
[0.13829787 0.65882353]
[0. 0.08235294]
[0.18085106 0.76470588]
[0.69148936 0.29411765]
[0.46808511 1. ]
[1. 0. ]
[0.21276596 0.09411765]
[0.41489362 0.91764706]]
"""
零均值标准化:或者叫均值标准差归一化;把所有数据都映射到均值为0标准差为1的分布中,这种归一化的方式则可以解决边界问题的。当然即便没有明显边界,比如学生的成绩等等,也是可以使用这种方法的。
\(x_{scale} = \frac{x - x_{mean}}{S}\)
import numpy as np
x = np.random.randint(0, 100, size=(10, 2))
print(x)
"""
[[17 65]
[25 39]
[80 88]
[ 3 50]
[17 26]
[77 67]
[22 94]
[19 78]
[28 88]
[98 16]]
"""
x_mean = np.mean(x, axis=0)
x_std = np.std(x, axis=0)
x = (x - x_mean) / x_std
print(x)
"""
[[-0.68738238 0.15019008]
[-0.43279631 -0.85107712]
[ 1.31748289 1.03592645]
[-1.13290799 -0.42746407]
[-0.68738238 -1.35171072]
[ 1.22201311 0.22721063]
[-0.52826609 1.26698811]
[-0.62373586 0.65082368]
[-0.33732654 1.03592645]
[ 1.89030153 -1.73681348]]
"""
数据没有明显的边界,原始数据可能存在极端数据值。
sklearn中的Scaler
对于哪些量纲不一致的数据集,我们是要进行归一化的,但是要如何体现在训练数据集和测试数据集上面呢。
很容易想到,对训练数据集求出mean_train和std_train,归一化之后训练模型。然后再对测试数据集求出mean_test和std_test,归一化之后进行预测不就可以了吗? 但是这样是不对的。
我们正确的做法是使用训练数据集的mean_train和std_train对测试数据集进行归一化。原因如下:
测试数据是模拟真实环境,真实环境下很有可能得不到所以测试数据的均值和方差对数据的归一化也是算法的一部分
因此我们需要保存训练数据集得到的均值与标准差。这在sklearn中也提供了相应的算法,而且被封装成了一个类,并且这和其它算法的格式是类似的,在sklearn中,所有算法都保证了格式的统一。
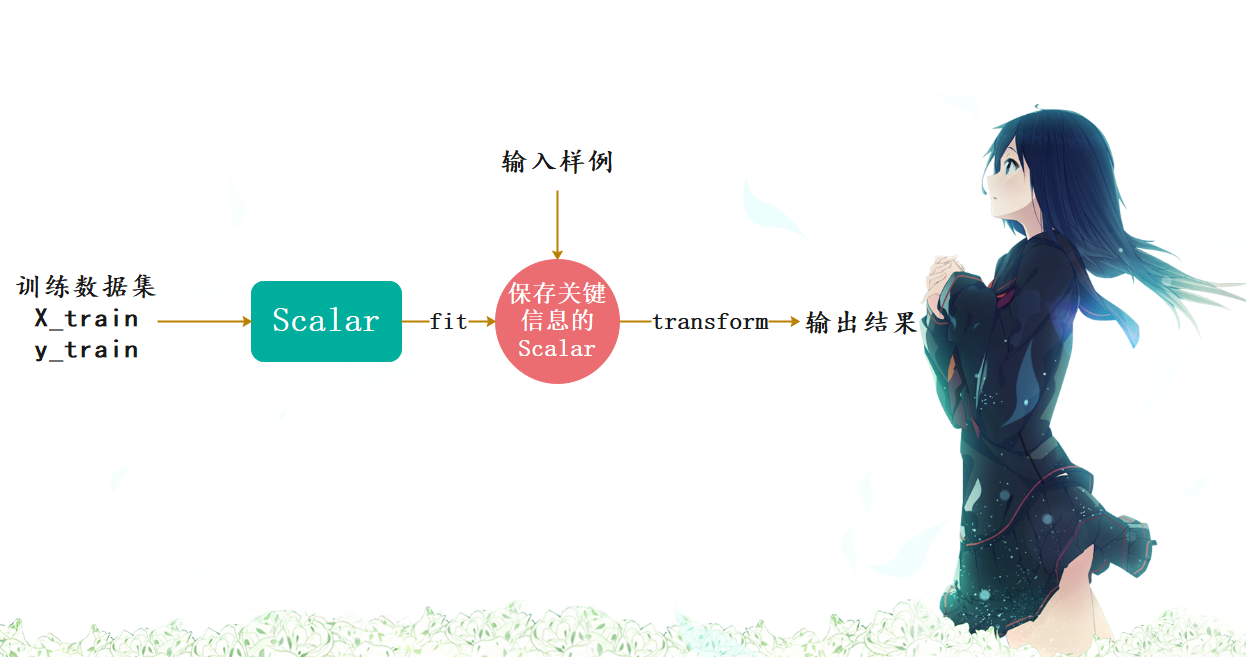
from sklearn.datasets import load_iris
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import StandardScaler
from sklearn.neighbors import KNeighborsClassifier
iris = load_iris()
X = iris.data
y = iris.target
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.3)
std_sca = StandardScaler()
# 这一步相当于计算出X_train中的mean和std
std_sca.fit(X_train)
# 查看均值和标准差
# 还记得这种结尾带下划线的参数吗?表示这种类型的参数不是我们传进去的,而是计算的时候生成的,还可以给我们后续使用的。
print(std_sca.mean_) # [5.78285714 3.0552381 3.62190476 1.14285714]
# 这里的scale_指的就是标准差
print(std_sca.scale_) # [0.78757701 0.45313321 1.73666242 0.74294871]
# 调用transform方法,对数据进行转化
scale_X_train = std_sca.transform(X_train)
scale_X_test = std_sca.transform(X_test)
knn_clf = KNeighborsClassifier()
# 对归一化之后的数据进行fit
knn_clf.fit(scale_X_train, y_train)
# 对归一化之后的数据进行打分
score = knn_clf.score(scale_X_test, y_test)
print(score) # 1.0
# 由于鸢尾花数据集比较规整,当我们进行数据归一化的时候,分类准确率达到了百分之百
# 但是注意的是,当我们对训练数据集进行归一化处理的时候,对测试集也必须要进行归一化处理,如果不进行归一化处理的话,那么结果肯定是不准确的。
# 我们可以尝试使用原始的测试集进行预测,看看结果如何
print(knn_clf.score(X_test, y_test)) # 0.3333333333333333
# 可以看到结果的分数只有0.333,显而易见如果只对训练集进行归一化处理而不对测试数据集进行归一化处理的话,那么结果会非常糟糕
那么我们能不能自己实现一个StandardScaler呢?显然是可以的,其实sklearn还有很多其他的归一化方式,比如我们之前提到的最值归一化,这在sklearn中被封装为MinMaxScaler,有兴趣可以自己尝试,我们这里介绍零均值标准化。
import numpy as np
from sklearn.datasets import load_iris
iris = load_iris()
X = iris.data
class MyStandardScaler:
def __init__(self):
# 这两个参数用户不需要传,而是根据训练自己生成
self.mean_ = None
self.scale_ = None
def fit(self, X):
# 根据训练集X得到均值和标准差
self.mean_ = np.mean(X, axis=0)
self.scale_ = np.std(X, axis=0)
return self
def transform(self, X):
assert self.mean_ is not None, "在transform之前必须先fit"
assert X.shape[1] == len(self.mean_)
return (X - self.mean_) / self.scale_
def __str__(self):
return f"{self.__name__}(mean_: {self.mean_}, scale_: {self.scale_})"
def __repr__(self):
return f"{self.__name__}(mean_: {self.mean_}, scale_: {self.scale_})"
from sklearn.preprocessing import StandardScaler
std_sca = StandardScaler()
my_std_sca = MyStandardScaler()
std_sca.fit(X)
my_std_sca.fit(X)
print(std_sca.mean_, std_sca.scale_)
# [5.84333333 3.05733333 3.758 1.19933333] [0.82530129 0.43441097 1.75940407 0.75969263]
print(my_std_sca.mean_, my_std_sca.scale_)
# [5.84333333 3.05733333 3.758 1.19933333] [0.82530129 0.43441097 1.75940407 0.75969263]
print((std_sca.transform(X) == my_std_sca.transform(X)).all()) # True
# 可以看到我们自己实现的归一化和sklearn提供的归一化得到的是一样的结果。
更多有关K近邻算法的思考
到目前为止,我们对K近邻算法算是有了一个清晰的认识了,那么K近邻算法都有哪些优点和缺点呢?
优点
K近邻算法用于解决分类问题,有些算法不适合解决分类问题,或者只适合解决二分类问题,而k近邻天然可以解决多分类问题。而且思想简单,功能强大。
不仅如此,K近邻还可以解决回归问题。回归问题和分类问题不同,分类问题是预测出一个类别,换句话说,种类是有限个。而回归问题,则是预测出一个具体的数值,数字有千千万万个,肯定不能用分类来解决。
那既然如此的话,k近邻又如何解决回归问题呢?
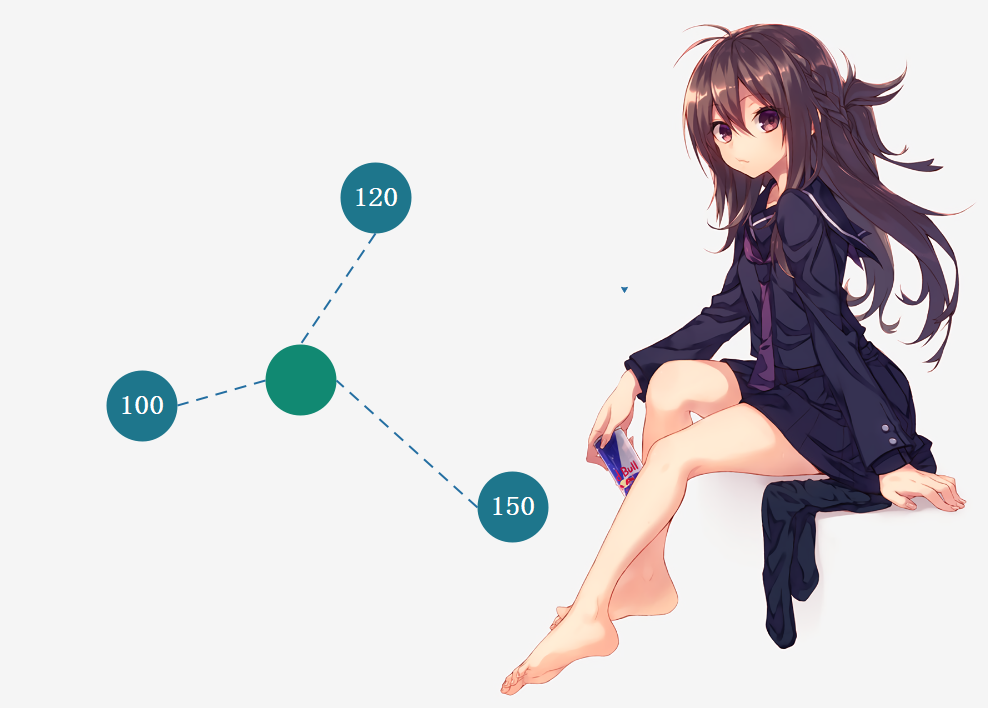
比如绿色的周围有三个点,那么K近邻可以取其平均值作为预测值,当然也可以考虑距离,采用加权平均的方式来计算。事实上sklearn不仅提供了KNeighborsClassifier(k近邻分类),还提供了KNeighborsRegressor(k近邻回归),关于回归会在后面介绍。
缺点
最大的缺点就是效率低下,如果训练集有m个样本,n个特征,则预测一个新的数据,时间复杂度是O(m*n)。因为要和样本的每一个维度进行相减,然后平方相加、开方(按照欧拉距离),然后和m个样本重复相同的操作。优化的话,可以使用树结构:KD-Tree,Ball-Tree,但即便如此,效率依旧不高。
第二个缺点是高度数据相关,理论上,所有的数据都是高度相关的,但是k近邻对数据更加敏感。尤其是数据边界,哪怕只有一两个数据很远,也足以让我们的预测结果变得不准确,即使存在大量的正确样本。
第三个缺点是预测结果不具有可解释性。
第四个缺点是维度灾难,随着维度的增加,看似相近的两个点之间的距离会越来越大
机器学习流程回顾
由于K近邻是我们学习的第一个算法,也是最简单的算法,所以我们把机器学习中的一些其它知识也放在这里介绍了。下面来回顾一下流程:

总结一下就是:
将数据集分成训练集和测试集对训练集进行归一化,然后训练模型对测试集使用同样的scaler(对测试训练的scaler)进行归一化,然后送到模型里面网格搜索,通过分类准确度来寻找一个最好的超参数
如果觉得文章对您有所帮助,可以请囊中羞涩的作者喝杯柠檬水,万分感谢,愿每一个来到这里的人都生活愉快,幸福美满。
微信赞赏

支付宝赞赏


 浙公网安备 33010602011771号
浙公网安备 33010602011771号