

edgeR
edgeR:Empirical Analysis of Digital Gene Expression Data in R
一个差异性分析的R包,用于RNA-seq或DNA甲基化等相关技术分析。
其原理利用广义线性模型对每个基因或者甲基化位点建模,然后直接比较线性模型的参数。
输入要求:必须是支持该位点的原始read count,而不是经过normalization计算的结果。
对于RNA-seq可以是htseq-count的结果。
对于甲基化分析可以是bismark的结果。
edgeR是以DEGList的格式储存数据,DEGList是一个以list为基础的数据格式,list的所有方法其都可以使用。
1.构建DEGList:用DEGList()构建。
>y<-DGEList(counts=x) #x是read counts的matrix或data.flame。 >group<-c(1,1,2,2) #关于sample属于哪一个group。 >y<-DGEList(counts=x,group=group)
DEGList中主要包括一个 $counts,一个 $samples ,还有一个可选的$genes (注释)。
$sample:
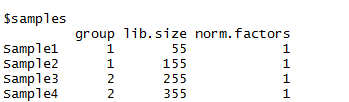
lib.size:默认为count的每一列总和,代表了该样品的总深度。
2。过滤:
生物学上看,一个基因要被表达成蛋白或是其他的生物功能,则它的表达量应该达到一个最低的水平。
所以在进一步分析前,应该过滤掉一些low counts的基因。这里用cpm(count per million)来表示基因的counts水平。
cpm的计算举例:比如在$count中一个点是10 ,该位点对应的sample的lib.size=50000.
该点经过cpm计算后得到的值(X):10/50000=X/1M
>keep<-rowSums(cpm(y)>1)>=2 #>=2表示每个group中的samples数最少是2. >y<-y[keep, ,keep.lib.sizes=FALSE]
3.TMM标准化:
在treated和untreated样品中,常常会有少量的基因在treated样品中高表达,但在untreated样品则正常。在treated样品中,高表达的基因的reads会占据一大部分的library size,
而导致剩余基因被错误的判断为下调。
>y<-calcNormFactors(y) >y$samples
4.设计矩阵(design matrix)
补充:表达式(~)
~0+group :不包括比较矩阵
~group:包括了比较矩阵
design<-model.matrix(~group)
5.离散度的检测(dispersion):
补充:
什么是离散度:
参考:
http://blog.sina.com.cn/s/blog_5d188bc40102vwci.html
http://blog.sciencenet.cn/blog-508298-776802.html

