7种测序平台
1. illumina测序化学原理
2. HiSeq 测序仪工作原理
3. PacBio 单分子超长测序
4. Ion Torrent 测序
5. X10人全基因组测序
6. illumina 公司的Nano Well 测序技术
7.第一代DNA测序
1. illumina测序化学原理
今天的第一期节目,主要给大家介绍Illumina公司的技术。Illumina公司是当今最红火的二代测序公司。它的测序技术的最基本的原理,是基于可逆终止的、荧光标记dNTP来做“边合成、边测序”的工作
听起来有点拗口,接下来逐步为大家讲解。
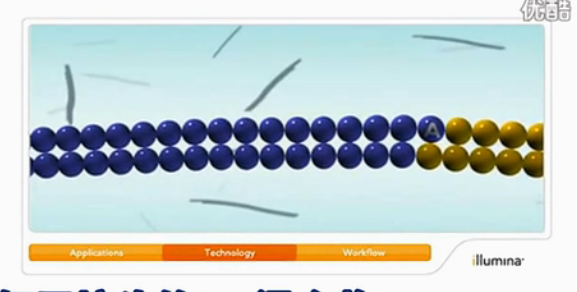
Flowcell

第一个要给大家讲的,是它这个flowcell。Flowcell翻成中文,就叫“流动池”。
我们来看这个图片。图片当中,我们看到一个象载玻片大小的芯片。这个芯片里面,是做了8条通道。在这个通道的内表面,是做了专门的化学修饰。它的化学修饰,主要是用2种DNA 引物,把它(2种DNA引物)种在玻璃表面。

这两种(DNA引物的)序列是和接下来要测序的DNA文库的接头序列相互补的。而且这2种引物是通过共价键,连到Flowcell上去。之所以要用共价键连到Flowcell上去,是因为接下来有大量的液体要流过这个Flowcell,只有有共价键连接的这些DNA,才不会被冲掉。
这就是Flowcell。
文库制作
再接下来,讲一下文库、和文库的制作(过程)
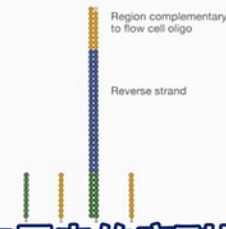
所谓的DNA文库,实际上是许多个DNA片段,在两头接上了特定的DNA接头,型成的DNA混合物。
文库有2个特点,第1个特点,是当中这一段插入的DNA,它的序列是各种各样的。第2个特点,它的两头的接头序列,是已知的,而且是人工特地加上去的。
要做这个文库,首先是把基因组DNA,用超声波打断。然后打断之后,两头用酶把它补平,再用Klenow酶在3’端加上一个A碱基。然后,再用连接酶把这个接头给连上去。


连好了接头的DNA混合物,我们就称为一个“文库”。英文也称作“library”。
桥式PCR

做好了Library之后,就要做桥式PCR了。桥式PCR,实际上是把文库种到芯片上去,然后进行扩增,这样的一个过程。
这个过程,首先是把文库加入到芯片上,因为文库两头的DNA序列,和芯片上引物是互补的,所以,就会产生互补杂交。


杂交完了之后,我们在这里面加入dNP和聚合酶。聚合酶会从引物开始,延着模板合成出一条全新的DNA链来。
新的这条链,和原来的序列是完全互补的。
接下来,我们再加入NaOH碱溶液。DNA双链在NaOH碱溶液存在下,就解链了。而且被液流一冲,原来的那个(模板)链,也就是没有和芯片共价连接的链,就被冲走了。而和芯片共价连接的链,就被保留下来。




然后,我们再在液流池里加入中性液体,主要是为了中和这个碱液,在加入中和液之后,整个环境变成中性了。这时侯,DNA链上的另外一端,就会和玻璃板上的第二种引物,发生互补杂交。
接下来,我们加入酶和dNTP,聚合酶就延着第二个引物,合成出一条新链来;然后,我们再加碱,把2条链解链解开;然后,我们再加中和液,这时侯,DNA链会和新的引物杂交。再加酶,再加dNTP,又从新引物合成出新的链来。
连续重复这一过程,DNA链的数量,就会以指数方式增长。



制备单链
在桥式PCR完成之后,接下来要做的工作,就是要把合成的双链,变成可以测序的单链。
办法是通过一个化学反应,把其中一个引物上的一个特定的基团给切断掉。
然后,再用碱溶液来洗这个芯片。这时侯,碱让DNA的双链解链,那根被切断了根的DNA链就被水冲掉了。留下那根共价键连在(芯片)上面的链。

接下来,再加入中性溶液,然后在这个中性溶液里面加入测序引物。
正式测序
好,接下来正式的测序工作就开始了。
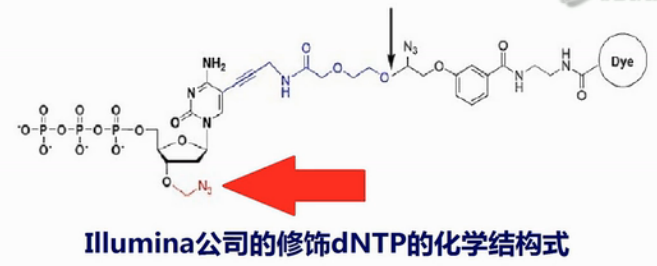
那么,在测序的时侯,加入进去的,最主要是2个东西:一个是带荧光标记的dNTP。而这个dNTP,它还有一个特点,它的3’末端是被一个叠氮基堵住的。
然后,再加一个聚合酶,聚合酶就会选择:哪一个dNTP是和原来位置上的那个碱基是互补的,根据互补性原理,把这个dNTP合成到新的这个DNA链上去。
因为这个dNTP的3’端是被一个叠氮基团堵住了,所以,它一个循环只能延长一个碱基。然后,它就停在那儿了。
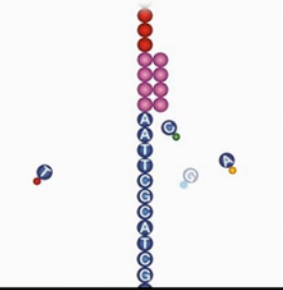

合成完了之后,就用水把多余的dNTP和酶给冲掉。
冲掉之后,就放到显微镜下,去进行激光扫描。根据发出来的荧光来判断它是哪个碱基。
因为4种dNTP,它每一种dNTP上面标的荧光素都不一样,根据红、黄、蓝、绿,它出来的哪种颜色,那么,就可以倒过来推出来,这个新合成上去的碱基,是哪种碱基。
因为新合成的碱基,是和原来位置(的碱基)是互补的,所以,又推出模板上那个碱基是哪个。
这一个循环完成之后,就加入一些化学试剂,把叠氮基团和旁边标记的荧光基团切掉。切完了之后,3’端的羟基就暴露出来。
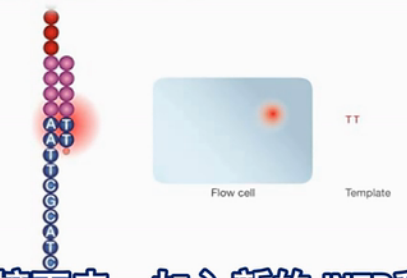
再接下来,加入新的dNTP和新的酶,然后,又延长一个碱基。新延长完一个碱基之后,把多余的酶和dNTP冲掉,再进行一轮显微的激光扫描,再读一下这个碱基是什么。
不断重复这个过程,可以重复上百次,到几百次,就可以把上百个碱基,甚至更多碱基的序列读出来。

读Index
那么,什么是Index哪?是因为Illumina的评委会个测序量很大,往往一个样本,用不了那么几亿条DNA。所以,科学家就想了一个办法。在文库的接头上做了一些标记,每一个样本,它有一个特定的接头,每个接头里面,它有一段特定的序列。
这段特定的序列,我们就称为Index。也有人把它叫做Barcode,反正,表达的是一个意思:这么一段特定的序列,标记了样本的来源。
那么,要读这个Index的序列,先用碱把上面这根测完“Read 1”的序列,把上面这根DNA链给解链掉。

解链掉之后,再加入中性液,然后,加入“Read 2”这个测序引物。Read 2测序引物结合的位点,正好,就在这个Index序列的旁边。
接下来,就进行第2轮测序,一般来说,是读6到8个碱基。把这6到8个碱基读下来,我们就可以知道,这某一个具体的一段DNA,它来自于原始的哪个样本。
双端测序

这是Illumina的最核心的另外一个技术,就是双端测序。
那么双端测序,就是说,一根DNA链,除了从正向读一遍,还可以从DNA的负向,再读一遍。
这一下子就把Illumina测序的有效长度加了一倍。这是非常有实际用途的。
那么这个倒链的过程,是这样,先让这个DNA先合成,合成出来这根互补链。
有了这个互补链之后,用一个化学试剂,在原来这根链的根上切一下。切一下,原来这根模板链就掉了,剩下那根互补链。
再接下来,就进行第2端的测序。第2端的测序原理,和第一端的测序原理是一样的。
加上了“Read 3”的这个引物,依次往下,一个一个碱基地往下读。
大规模平行测序

那么最重要的事情是什么呢?一个点,经过几百个循环,就读出了几百个碱基。但实际上,这个芯片上可以有上亿个点,上亿个“cluster”,也就是“簇”。那么上亿个“cluster”,每个循环,它都可以读出地么多序列,这是Illumina测序非常强大的原因。因为是成千上万,准确说是上亿上链都在合成,这个就得到了很大的一个测序数据量。
2. HiSeq 测序仪工作原理
上期节目,给大家介绍了Illumina测序的化学原理。这期节目,主要给大家介绍一下,Illumina HiSeq测序仪的工作原理。
也就是芯片上发生了这么多变化,HiSeq是如何把这些信息给读出来,并且把扫描出来的荧光信号,又通过怎样一系列的加工,变成可以识别的“A、C、G、T”的碱基序列的。

HiSeq首先是一台高精度的显微光学扫描仪。然后再配上了一整套的液流系统,和计算机软硬件,再加温控系统,组成这样一台测序仪。
其中最核心,也是结构最复杂的,是它的光学系统。
前一期,我们讲了,Illumina测序仪主要是靠4种dNTP分别带有不同的荧光基团,在被激光照了之后,发出不同颜色的荧光。再通过对光的颜色的分辩,可以判断出到底是哪个碱基。
光路结构
我们先来说,它的光路结构。
这里,我们要说明一下:感光元件CCD,它本身是色盲。所以,它一定要配合滤光片,才能分辩出颜色来。
那我们先来看一下,HiSeq的光路图。
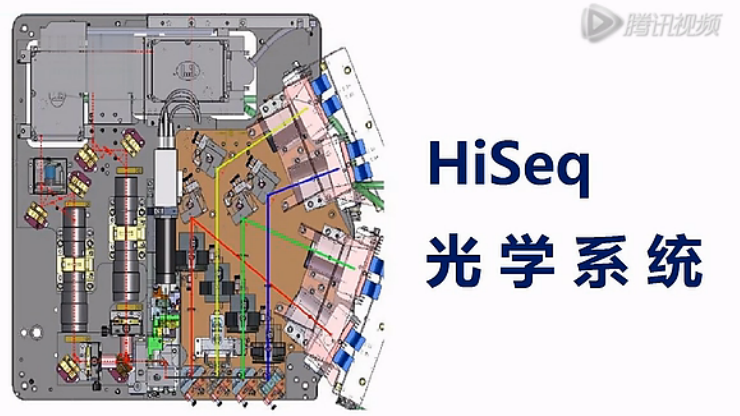
左边这两个元器件,就是激光器。一个发出红色激光,另一个发出绿色激光。
其中红色激光主要是激发A和C,这两种碱基上的荧光基团;而绿色激光主要是激发G和T,这两种碱基上的荧光基团。
红色和绿色这两束光,通过一面半透半反镜,组成一道激光。这道激光打在Flowcell上。

那么请注意,Flowcell就放在这个位置。
在Flowcell里面,结合在DNA上的那个荧光基团在激光的照射下,就发出荧光。
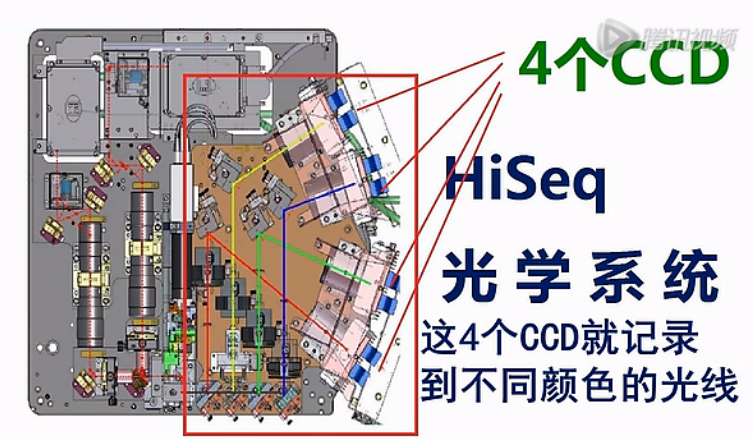
荧光通过3面半透半反镜,和1面全反镜,被分成4条光路,这4道光线,分别通过一道滤光片,这4张滤光片的滤过波长不一样。这样,这4 道光在经过了滤光片之后,就变成了4种颜色不同的光线。
然后,这4条颜色不同的光线,各自照在一面反射镜上,通过反射镜进入到CCD。这4个CCD就记录到不同颜色的光线。
TDI线扫描
HiSeq的光线扫描是“线扫描”,和传统的相机不一样,传统的相机是面扫描。
HiSeq采取了一种特定的叫“TDI”线扫描方式,TDI是Time delayintegration的缩写。
在HiSeq上之所以采取TDI扫描方式,因为它有非常明显的优点。
第一个优点,就是它的扫描速度非常快,在HiSeq 2500上,从Flowcell的一个Lane的一头扫到另外一头,也就是一个“Swath”的扫描时间,大概只有20秒种不到。
第二个好处,就是它的扫描精度非常高。在最新的HiSeq V4版试剂上,它的光点密度,大概可以达到每平方毫米90万个点,要扫描清楚这么高密度的光点,扫描仪的扫描精度是可想而知的。
TDI扫描的第三个好处,是这种方式,可以把Flowcell的上表面、和下表面都扫描到。
Flowcell(测序芯片)
接下来,我们再要详细介绍这张Flowcell。

那么,先来看一下,这张flowcell有点象一张载玻片,在这一张片子里面,我们可以看到,
它做了8条通道。
每条通道,我们称为一个Lane。这8个Lane之间,相互是隔绝的。
每个Lane的两端各有一个小孔。这两个小也孔,就是液流流进、流出的地方。

每个Lane的上表面和下表面,都分别以共价键的方式,种了2种DNA引物。这两种DNA引物,是与文库接头的两头序列相互补的。上一期(节目)我们已经说明了这一点。

一个Lane里面,分成2个面,上表面、和下表面。上表面和下表面,都种了DNA引物,也都是可以产生测序数据的。
在每一条Lane的每一个面,又被分成了3个扫描通道,每个道被称为一个“swath”。

每条Swath是从头到底被连续扫描的。但是它的数据,在进行数据分析的时侯,是被分割成16个小方块。这每一个小方块,被称为一个“tile”。
这样一张Flowcell,总共就是768个Tile。
每个Tile在扫描的时侯,会根据4种颜色,产生4张照片。

图像处理
扫描完了之后,就要进行图像处理。
扫描出来的最原始的文件,它的格式是“.tiff”文件。
Tiff文件记录了每个像素点上采集到的光强度。Tiff文件的优点是它是完全无损,保留了所有的原始信息。
但它也有它的不足之处。它的不足之处就是它的这个文件太大了。它的数据量很大,既不便于数据的传输,也不便于数据的存储。
接下来,计算机软件就把图像文件转化成光点文件。光点文件叫“.BCL”文件。也就是“Base calling”的英文缩写。
要把图像文件,转化成BCL文件,就是把4种颜色的4张照片,组合在一起,变成一张有4种颜色的彩色照片。

这其中首先要解决的,是4张照片在空间位置上的匹配问题,因为4张照片是通过4个CCD分别拍下来的,所以,会有一定的空间上的偏差。
软件要通过对4张照片上,亮点相互比对,找到最合适的、匹配的位置。
这里,我们要说明一下,如果被测的文库是碱基不平衡的文库,在这个空间匹配上就会遇到问题。
什么叫碱基平衡呢?也就是说,在测序过程当中,每个循环,A、C、G、T四种碱基,都是比较均匀在存在的。
最典型是人全基因组文库,这是一个典型的碱基平衡文库。
那什么是碱基不平衡文库呢?最典型的,就是PCR扩增子产生的文库。PCR扩增子的特点:PCR是有特定的起始位点的,一个特定的测序循环中,几乎所有的片段都是同一种碱基,而剩下的3种碱基,就特别少。
这在反映到照片上去的时侯,就变成:一张照片特别亮,光点很多。而其它的三张照片就特别暗,上面的光点就很少。
这时侯,要软件做空间上的比对,软件就会觉得困难,因为对于那几张暗的照片,软件很难判断上面的光点,是否与那张亮的照片上的光点真正对得上。结果,就是判断出来的可靠性变差。最后,就是测序的数据质量变差,有效数据量也会变少。
要解决这个问题,办法是在测序过程中掺入一些碱基平衡的文库。例如掺人全基因组文库。或者也可以掺Illumina提供的标准的PhiX文库,这些都是碱基平衡文库。
它的作用,是在每个循环当中,为每一种颜色的照片,都提供足够多的亮点。这样,它可以弥补那些不平衡的文库当中缺亮点的问题。
BCL文件
当把4种颜色的光点组成一个文件之后,软件就会生成一个“.BCL”文件。
“.BCL”文件就是光点文件,它对每个光点,记录了以下的内容。

首先一个光点处在哪个Lane里面。其次,这个光点在这个Lane的哪个Tile里面。第3,就是这个亮点在这个Tile的X轴和Y轴的座标位置。第4,是记录了这个光点当中“红、黄、蓝、绿”四种光的对应的光强。
这个图是BCL文件的一个示意图。
实际上,BCL文件是二进制文件,无法拿来直接阅读。也正是因为BCL文件难于阅读,并且很难改动,所以,BCL文件几乎不存在做假的可能。
在测序过程当中,有许多客户会要求测序公司提供原始的测序数据,如果客户是包Lane、或者包Flowcell的,一般测序公司是可以提供BCL文件的。
客户在拿到BCL文件之后,可以用“BCL2FASTQ”这个软件,把BCL文件转化成FASTQ序列语文件。
以此,客户可以来验证,测序公司提供的数据是否是原始的,是否是真实的。
再说一下最初生成的那个tiff文件。tiff文件实在太大了,所以,测序仪在测序过程中,只把tiff文件作为中间文件。最后是把这个tiff文件删掉的。
如果客户想要原始的图像文件,在HiSeq V4之前,可以让测序公司保留“.CIF”文件。CIF文件是一种彩色图案的向量文件,它的优点是比tiff文件的数据量小很多。
测序公司把CIF文件给客户之后,客户就可以看到原始的图像文件了。
但是,请注意:在HiSeq升级到V4之后,保留CIF文件的这个选项是被取消掉了。所以,对于要测V4 Lane的客户来说,是拿不到CIF文件了。
碱基识别
接下来,我们讲一下碱基识别。
我们之前讲:4种dNTP,各标一种荧光基团,红、黄、蓝、绿,四种颜色,根据颜色来判断碱基种类。这个实际上是一种简化了的说法。实际情况,要比这个复杂得多。
来看这个图,这是2种荧素的荧光的波长图。

我们会发觉,这两种荧光色,它发出来的发射光,它在波长上是有交叠的。在X的这个位置,主要是绿色荧光素的贡献,但是蓝色荧光素,也有少许贡献。
而在Y这个波长位置,蓝色荧光素是做了主要贡献,但是绿色荧光素,也有少量供献。
在实际测序过程中,是4种荧光素发出的亮,相互有交叠,相互之间的交系,变得更加复杂。

那么,现在我们要做的事情,是把A、C、G、T,4种荧光素的贡献给拆开。
首先,我们就要确定4种荧光素在4个被测波长处的贡献率。
我们可以看一下,这个表,就是4种荧光素,在4个波长分别有不同的贡献率。这样就组成一个4X4的贡献率表格。
我们在实际的分析当中,等于解一个4元1次、4联方程。因为是4个未知数,又是4个方程,所以肯定是可以解出来的。

说解方程,有点复杂。那么我们来打一个比方。让大家来理解这个事情。
假设有一家饭店,它有4个熟客:甲、乙、丙、丁。它日常又提供4道菜:猪肉、白菜、黄瓜、花生。大厨知道:甲最爱吃猪肉、乙最爱吃白菜、丙最爱吃黄瓜、丁最爱吃花生,每个人来了饭店之后,主要吃自己最爱吃的,也会吃些别的菜,但别的菜都吃得不是太多。

那么这个大厨不到前台,看不到今天来的客人。如果,这个大厨想要知道今天来的客人是谁,他有什么办法呢?看今天哪个菜被吃掉得最多。如果今天的菜被吃掉的最多的是猪肉,那他可以大致地判断,今天是甲来过了;如果他看到今天被吃掉的菜,最多的是白菜,很可能是乙来过了;那么其它的,道理也是一样的。希望这个例子可以帮大家来理解一下,这4个荧光和4种碱基的判读的关系。
Phasing 和 Prephasing
接下来,我们再讲一下,Phasing和Prephasing。
在Illumina的测序过程当中,一个簇,大概有5千个到1万个分子。但是在边合成、边测序的过程当中,每一步酶反应,理想情况下,应该这5千个分子都延长1个碱基。
但实际情况,总有少量分子没有完成延长反应。也就是说,总有少量的分子会掉队,我们称这种掉队的现象叫“phasing”。Phasing主要是由于酶活性不足,所引起的。

如图所示,掉队的这个分子,它所发出的荧光信号,和大部队所发出的荧光信号是不一样的。这个循环的次数越多,掉队的分子就越多。所以,测序越到后面,它Phasing的分子数就越多。最后,信号的可靠性就越差。
除了掉队的分子,还会有一部分分子,会跑得超前,也就是在一个循环中,它延长了2个碱基。在一个循环中延长了2个碱基的最主要的原因,是dNTP上标记的那个叠氮基团(N3)掉了。

我们知道,叠氮基团是非常容易从有机化合物上掉落的。当叠氮基团掉落之后,dNTP的3’端的羟基就暴露出来了。当丢失了叠氮基团的dNTP加到(合成链的)3’端之后,它的聚合反应不会终止,而是会继续往前走。当再加上了一个带叠氮基团的dNTP之后,这个聚合反应才停下来。
这样的后果,就是一个循环,某些分子,会合成了2个碱基。也就是说比大部队多走了一步。那么这个多走了一步的碱基,它所发出来的荧光颜色,也是和大部队不一样的。
在Illumina测序过程当中,Phasing和Prephasing是限制测长的最主要原因。也就是说,随着循环不断进行,越来越多的分子掉队,还有越来越多的分子超前。然后,它们所产生的噪音,掩盖了大部队的信号的时侯,也就是测序开始测不准的时侯。
在HiSeq测序当中,从第12个循环开始,在计算某个光点是哪种碱基的时侯,就要把Phasing和Prephasing的影响,纳入考虑。
Chastity 和 Pass filter

为了对光点当中荧光素的纯粹程度进行描述,Illumina公司定义了个标准,叫“chastity”,Chastity的定义,就是浓度最高的那个荧光素的量,去除以“它自己 + 排名第二的荧光素的量的和”。大于0.6是一个好碱基。
用更加通俗的话来说,也就是“老大”比“老二”,如果大于、等于“1.5倍”,这就是个“好”碱基。如果“老大”比“老二”不足“1.5倍”,这就是个“坏碱基”。
Illumina对每个read的质量都要做一个检验,这个检验就叫“pass filter”检验。检验的标准,是看前25个碱基当中,有几个是“坏碱基”。如果只有一个、或者没有坏碱基,则Pass filter就通过;如果有超过一个以上的坏碱基,Pass filter就不能通过。
那我们平时说,测序服务保证多少“PF data”,指的就是Pass Filter(PF)的数据。
Pass Filter最主要的作用,就是把那些一个光点当中,含了几个cluster的那些点,给去掉。只剩下那些纯粹的单克隆的read,作为合格的数据,提交给客户。

我们平时说“PF率”,指的就是Pass Filter的Reads数,占总的、测到的Reads数的比例。
PF率可以从一个侧面反映测序的质量。一般来说,如果上样密度过高,PF率就可能会下降。
Quality Score,Q 值
一个碱基的Quality Score,也就是这个碱基的质量分数(Q值)。这个是通过这个碱基被误判的可能性,换算出以10为底的对数,再乘以“-10”得到的这样一个数字。

这个Q值,有点象我们说黄金的纯度,我们说“三九金”,或者说“四九金”,就是指99.9%的纯度的金子,或者是99.99%的纯度的金子。
我们平时说Q30,就是指一个碱基的可靠性达到99.9%。或者说,它的出错的可能性小于千分之一。
同样道理,我们说Q40,就是指一个碱基的可靠性是99.99%。或者说,它的出错的可能性是万分之一。
那么,我们经常说Q30比例,所谓的“Q30比例”,就是在全部PF数据当中,达到、或者超过Q30质量标准以上的数据,占所有PF数据的比例,叫Q30比例。
Q30比例,可以表征一个测序过程的质量的好坏。一个碱基的质量分数,不是以数字方式,直接记录到最后的Fastq文件的。而是把它的Q值,加上33,再用ASCII码表转换成一个字母,把这个字母录入Fastq文件。

这样做,有2个好处。如果我记2位数字,那么就占2个字节,现在用一个字母来记录,只占一个字节。那(数据存储)空间就节省了很多。
第二个好处,用ASCII码字母表,一个碱基,只对应一个字母;如果是用2位数字来记录,就有可能发生移码错误。而用ASCII码,一个字母来记录,就不太容易发生移码错误。

Fastq 文件

在软件做完上述所有的数据处理之后,就会生成一个Fastq文件。
Fastq文件里,主要包含了3部分内容。
第一个部分,是每个Read的目录信息。也就是这个Read来自于哪台HiSeq、第几个run、第几个Lane、和第几个Tile,以及在这个Tile的X、Y的什么位置。
接下来,就是所测到的碱基的序列。
最后,是这些碱基序列对应的质量分数信息。
这个,就是Fastq文件。
到Fastq文件之后,测序仪所要完成的工作,就完全完成了。
以上,就是HiSeq测序仪的基本工作原理。
3. PacBio 单分子超长测序
本期节目,给大家介绍一下Pacific Biosciences公司的技术。Pacific Biosciences公司的简称叫PacBio。Pacbio是目前读长最长的测序技术公司。
它的读长,最长可以达到2万到3万个碱基,平均可以达到8千多个碱基。相比于llumina 和Ion Torrent的几百个碱基的读长来说,有着明显的优势。
今天,我们就给大家介绍一下这个技术。
PacBio 测序过程
PacBio的测序原理,和别的高通量测序的原理,基本上也是一样的。也是边合成,边测序。
首先,这个聚合酶是固定在测序小孔的玻璃底板上。这个聚合酶又和DNA模板、测序引物是结合在一起的。


然后加入带4色荧光的dNTP底物,这些dNTP都在其磷酸基团上被标上了荧光基团,四种碱基、各标一种颜色。
当一种与聚合酶正要合成的碱基一致的dNTP被酶抓住的时候,酶就会长时间地抓住这个dNTP,不让这个dNTP漂走。
这时侯,激发光从小孔的底部照进来,打在这个被抓住的dNTP上,就会在较长时间内发出荧光。

仪器根据所拍到的荧光的颜色,就可以来判断,这个碱基是哪种碱基。
一个循环的聚合反应发生完毕之后,焦磷酸基团就从原来的dNTP上掉下来,因为荧光基团是连到这个焦磷酸上的,所以这个荧光基团也就一起掉下来了,在溶液中就会漂走。
接下来,进行第二、第三个循环……,一直进行下去。
一张芯片上有几万个孔,同时进行测序,这样一次就可以得到几亿个碱基的序列。

接下来,分几个要点,来说明这个测序的过程。
化学方法
和Illumina一样,PacBio也采用了4色荧光基团来标记dNTP,但是PacBio的标记和Illumina的标记有所不同,PacBio的荧光基团直接是标在dNTP的3'端的磷酸基团的末端的。
这样标记的好处是:当一个聚合反应的循环完成的时侯,dNTP上的那两个磷酸基团就掉下,连在这个磷酸基团上的荧光基团也随一块儿掉下来。它掉下来之后,就在溶液中漂走,不会影响接下来的测序过程了。
测序微孔
然后,我们说一下这个测序小孔的设计。

这个测序小孔叫Zero Model Waveguide,简称ZMW。
小孔的直径很小,光只能在小孔中传输很短的距离。这个特点对PacBio的测序很重要。因为酶是被固定在玻璃底板上的,所以,只有互补的dNTP被酶抓到的时侯,这个dNTP才会较长时间地停留在离玻璃底板很近的位置。
也只有这样,才会被激发光照到,并且发出它的荧光。

PacBio的光学设计中,入射光是几百纳米波长的可见光,光从小孔的底部的玻璃处照到小孔中来。这个,只有70纳米。
其它游离的dNTP,只会非常短暂地进入小孔,又很快漂走。所以,这些游离dNTP带来的的噪音(信号),就被抑制在很低的水平。
哑铃状的文库


接下来,我们说一下PacBio的建库。
PacBio的建库是比较特别的。它的库是在DNA片段的两段各接一下发夹型的接头。接好了发夹形的接头之后,形成的文库是一个哑铃形的文库。

这种哑铃形状的文库有个好处,那它整个分子实际上是一个圆环。在测序的过程中它可以周而复始地进行测序,这对于发挥PacBio的长读长的优势是很有益处的。
超长读长的根本原因 -- 单分子测序
接下来,我们说一下PacBio它测序长度优势的来源。这个来源,是因为它测的是个单个分子。
相比之下,Illumina或者Ion Torrent测的都是一簇分子。或者说它们测的都是一大堆分子。当它测一大堆分子的时侯,每个循环,多多少少,总有一些分子落后;也多多少少,有些分子超前。
这些落后、或者超前的分子,在每个循环里面就会给出噪音。而且,随着循环次数越来越多,落后、和超前的分子也会越来越多,达到一定程度的时侯,噪音就会很大,大到会掩盖掉信号。
当噪音大到掩盖掉信号的时侯,实际上测序就测不准了。
相比之下,PacBio它只有一个分子,所以,它不存在同步问题。
这就让它可以测到几千、基至上万个BP都可以达成。
碱基判读准确率:87.5%
接下来,我们要说一下PacBio测序的缺点。
最大的缺点是对碱基的判读不准。它的错误率是12.5%。也就是说,它每读8个碱基,就有一个是读错的。
那么它主要的错误类型是"插入"。也就是说,它会多读一个碱基。
好在,它的这种错误是随机的。也就是说,你在这个地方再读一遍,它不一定会发生同样的错误。那么,对于同一个序列,多测几遍之后,这些偶然误差,可以被校正过来。
读长限制因素
接下来,我们说一下限制PacBio读长的因素。
第一个因素,就是DNA链上出现了缺口。测序过程中是用激光照射来发出荧光的,所以当强光长时间照射DNA链的时侯,DNA链就有可能被照断掉,出现缺口。
当酶读到这个缺口的时侯,酶就从模板链上掉下来。这时侯,测序就终止了。这是第一种可能。
第二种可能,是光线照射情况下,酶有可能会变性,当酶发生了变性之后,失去了聚合酶的功能,这时侯,测序也会终止。
第三个限制因素,是文库本身的长度。因为要做片段长度大于20~30K的文库,是有相当大的困难的,所以,文库本身的质量,在一定程度上,也限制了PacBio的读长。
数据通量
在高通量测序当中,测序的通量,是一个很重要的技术指标。
那PacBio大根一张芯片一次可以测到0.3~0.4G的数据。
在PacBio测序中,芯片上的小孔数是第一个绝对的、限制性的因素。
目前的芯片,是有15万个小孔。
但这15万个小孔中,并不是每一个都能产生有效数据的。
这里,我们要说一下,测序复合物和玻璃底板结合的方式。
所谓的测序复合物,就是"聚合酶、测序模板、测序引物"这三者组成的复合物。
这个复合物是通过聚合酶连接到玻璃底板上的。
这个连接方式,首先在聚合酶上标上生物素。然后,在小孔的玻璃底板上标上链霉亲合素。
实验过程当中,利用生物素和链霉亲合素的亲合力,把两者(聚合酶、和玻璃底板)结合到一块儿。
在实验过程当中,这个测序复合物是被随机地铺撒到这15万个小孔中的。因为是随机地铺撒进去的,所以,有多少个小孔里面正好有一个测序复合物,是符合泊松分布的。
最理想的情况下,是有1/3的小孔是正好有一个测序复合物。这时侯,还有约1/3的小孔是空的,还剩下约1/3的小孔是有2个或者3个以上的测序复合物被种进去。
空的这些小孔,因为接下来它没有聚合反应发生,也没有信号,那当然是废掉了。
那么有2个复合物种进去、或者有更多复合物种进去的这些小孔,因为它产生的信号会非常的杂乱,所以,这些孔实际上也是没用的。它产生的数据,在接下来的数据分析当中,是会被去掉的。
一张芯片有15万个孔,其中1/3有效,也就是说,有效的孔数是5万个。然后乘以它目前的平均测长,大概8千多个碱基,所以,一张芯片,比较理想的情况下,大概有0.4G的数据量的产出。
直接测DNA修饰
PacBio在测序当中,可以直接测到碱基的被修饰状态。因为当聚合物,遇到模板上有甲基化的A、C等碱基,它测序的速度就明显地放慢。而且它的光谱特征会发生改变。这样,就可以判断,这个位置上的DNA被甲基化了。

GC Bias 很小
PacBio测序还有另外一个好处,就是它GC Bias很小。
什么叫GC Bias呢?就是我们知道,所有的PCR的过程,如果模板里面G、C(碱基)的含量比较高,PCR的效率就比较低。反之,A、T(碱基)的比例比较高,则它PCR的效率比较高。
传统的建库当中,一般都有大量的PCR的过程。它导致的一个结果,就是G、C含量高的那些片段,它读到的Reads数,就会比较少。
PacBio它的好处,就是它的建库过程中没有PCR过程,所以,它带来一个直接的好处:就是它测序过程当中,GC Bias很少。也就是说,那些高GC的片段,有和低GC的片段差不多的概率被读到。

测序速度极快
高通量测序的另外一个指标,就是测序的速度。PacBio的测序速度取决于酶反应的速度。
目前PacBio用的这个酶,大概1秒钟是合成3个碱基,1个小时大概就可以合成1万多个碱基,3个小时可以合成3万多个碱基。到3万多个碱基之后,基本上继续在读的Reads,已经几乎没有了,所以,3个小时之后,测序基本就完成了。
1个Run读三个小时,相对于Illumina的测序速度来说,是非常快的;相对于Ion Torrent的测序速度来说,也相对要快一点点。
所以,PacBio是一种非常快的测序方式。
国内现有PacBio测序仪的单位
最后,把国内已经采购了PacBio测序仪的单位,列个清单,以方便大家相互合作。
-
天津市湖滨盘古基因科学发展有限公司,4台PacBio. http://www.pangugene.com/
-
北京遗传所,1台PacBio
-
北京药植所,1台PacBio
-
上海南方基因中心,1台PacBio
-
云南动物所,1台PacBio
-
武汉邓子新教授实验室,1台PacBio
-
天津生物芯片中心,1台PacBio
-
海克维尔基因(北京)研究院,1台PacBio
-
北京华牛世纪生物技术研究院,1台PacBio
-
上海交通大学 分析测试中心,1台PacBio
-
内蒙古农业大学,1台PacBio
-
华大基因,1台PacBio
4. Ion Torrent 测序
今天,要给大家介绍的是Thermo Life公司的Ion Torrent测序平台的测序原理。
我们分成以下4个部分来介绍Ion Torrent的测序原理。
-
建库过程
-
文库通过油包水PCR过程,变成可以上机的珠子的过程
-
上机测序过程
-
Ion Torrent测序平台的优缺点
1、建库过程
那么,我们先来介绍一下建库过程。
建库是在样本DNA片段的两侧加上标准的接头的这样一个过程。
Ion Torrent的建库中,接头是平头的,这点是与Illumina文库的接头是3’端有带突出的T碱基粘性末端的是不同的。
在加接头的过程当中,是加入P1接头,并同时加入X接头或者A接头。
其中,X或A接头是未来的测序起始端。而P1接头是是连到测序珠子的这一端。
X接头和A接头的差别是:X接头是带Barcode序列的,而A接头是不带Barcode序列的。用X接头的好处是可以把一个芯片的测序通量分配个几个文库,测完序之后用Barcode把不同的文库的序列给分开。A接头的好处是直接测到样本序列,这样对于充分利用测序的读长是更好的.但是它的缺点是没有Barcode,所以一张芯片只能放一个样本。

在Ion Torrent测序当中,AmpliSeq文库是很一种非常常见的文库。AmpliSeq文库是通过多重PCR扩增出来的DNA,再加上接头,做的文库。
如果把整个的PCR扩增产物都拿来测序,那么测到的两头20~30个碱基,都会是PCR引物的序列。而PCR引物是人工设计的,它的序列是已知的。如果把这些PCR引物都进行测序,那就会浪费相当大的一个测序读长和测序数据量。那为了解决这个问题,Thermo Life公司在设计Ampliseq的PCR引物的时侯,在这个引物上特别设计了一种化学修饰,这种化学修饰可以被Fupa(视频中写作Pufa,正确的该是Fupa)试剂所切断。这样,利用Fupa试剂把PCR扩增产物上大部分的引物序列都给切掉。在测序的时侯,就可以尽可能多地测到样本序列。



这是一个非常巧妙的设计。
2、油包水PCR
在做好文库之后,接着就要做第二步:就是把文库种到测序珠子上去,并且进行扩增。
Ion Torrent把文库种到测序珠子的方法,是做油包水PCR。也叫EmulsionPCR(乳浊液PCR)。油包水PCR包括两个相:油相和水相。其中水相是核心,油相起到分隔作用。水相中包括文库、引物、酶、Master Mix、测序珠子,这5种PCR反应的主要成份。
其中这个测序珠子,它是接下来测序的核心载体。这个测序珠子是非常小的,PGM测序仪上用的珠子,它的直径大概平均是2.4微米;Proton PI测序芯片上用的珠子,它的直径,大约只有一个微米。这些测序珠子的表面,共价连接了许多PCR引物,这个引物的序列正好是和前面文库的P1接头是互补的。

每一个油包水PCR都会包含许许多多个这样的、小的测序微珠。
水相中的另外一个成份是那个游离的PCR引物。这个游离的PCR引物,它的5’端标记了生物素。这个标记的生物素的作用,我们后面还要讲到。
这个引物的序列,是和前面的A接头、或者X接头相一致的。
准备水相,就是把文库、引物、酶、Master Mix、测序珠子先在水相中混合好。
混合好之后,加入油。把油和水进行混合,形成乳浊液。在这个乳浊液当中,大部分是油。油把水相分隔成一个一个的小水滴。
每个小水滴当中都可能含有0个到若干个文库分子,还会包含0个到若干个的小的测序微珠。与此同时,引物、酶、和dNTP是过量的,所以几乎每个小水滴中都会有足够量的引物、酶、和dNTP。在整个油包水PCR反应当中,文库分子和测序微珠是限量因素。

接着,把混合好的乳浊液,进行PCR反应。
PCR反应的结果是,在一个小水滴当中,如果它同时有文库分子、和测序微珠,它就会发生PCR反应。如果缺少了文库分子、或者测序微珠,它就不会发生PCR反应。
PCR反应之后,珠子的表面,就会长出以同一个液滴当中,所含的DNA(文库)分子的扩增拷贝来。这些扩增出来的DNA链,是通过共价键连到珠子上的,这个共价连接,可以保证在接下来的测序过程当中,这些连到珠子上的DNA链,不会被(液流)冲走,那么这些DNA链就可以作为稳定的测序模板。
油包水PCR完成之后,要把所有珠子当中,那发生了PCR的珠子给纯化出来。
纯化的手段,是通过用标记了链霉亲合素的磁珠,和刚才经过PCR扩增反应的珠子进行混合。那些发生了PCR反应的珠子,它上面的DNA链,是连了一个从PCR扩增引物中带的生物素的,生物素会和链霉亲合素很牢固地结合。这样磁珠就会和发生了PCR反应的测序珠子结合在一块儿;而那些没有发生PCR反应的珠子,它上面没有连着生物素,所以,它不会和磁珠结合。
接下来,我们用磁铁来进行吸附,磁铁会吸附磁珠,磁珠会把带了生物素、同时带了扩增了的DNA链的那些测序珠子给富集起来,而那些没有和磁珠结合的那些微珠,是留在上清液当中的,通过清洗,就被洗掉了。
然后,通过专门的洗脱液,把磁珠所富集起来的测序珠子给洗脱下来。
3、上机测序
这些洗脱下来的珠子,就可以上机测序了。
第3部分,我们来介绍Ion Torrent的上机测序。


Ion Torrent测序芯片,是一人半导体芯片,上面做了数以百万、千万计的小孔。它每个小孔的既是测序微珠的容器,又同时是一个微型的PH计。每个小孔正好可以容纳一个测序微珠。在测序过程中就是通过测量、并记录每个小孔中,所发生的PH值的变化,来测出DNA的序列的。

每个芯片,都会有一个进口,还会有一个出口,是走液流用的。把测序珠子上机的时侯,就从进口把珠子的混悬液注入到芯片上。然后再把这个芯片离心一下,离心的作用,是更好地把珠子卡到小孔中去。
Ion Torrent的测序原理是:每个dNTP分子都有3个磷酸基团,当dNTP被聚合酶结合到DNA链上时,会掉下来的一分子的焦磷酸,1个焦磷酸分子会被酶再进一步分解成2个磷酸分子,这样,在测序的微环境中,就会多出两个酸性分子,一个珠子上有几千、几百条DNA链,每次发生聚合反应,就会多出几千、几百个酸分子。这样,这个微环境的PH值就会短暂地下降。

这时,Ion Torrent芯片中,每一个小孔中的PH电极,就测量这个小孔中的PH值变化,并且把测量得到的值传给计算机。
分别含A、C、G、T四种dNTP的溶液,分别地、依次地流过芯片的表面。
举例来说,流入的是dCTP溶液,而模板上正好有一个G碱基,就发生聚合反应,并产生电压变化,而且会被记录下来。如果流入的溶液与模板上的碱基不匹配,就不会发生聚合反应,也就没有电压变化,也就不会有碱基被记录下来。
如果正好有2个一样的碱基相邻,一次就会有2个碱基被聚合到DNA链上,电压变化值就会加倍,序列中2个新的碱基被记录下来。

在测序的序列安排上,最前面的4个碱基叫Key sequence,分别是A、C、G、T。因为每个珠子上长多少个DNA链,它的变化范围是很大的,所以用Key Sequence的A/C/G/T四个碱基所测到的PH值变化的强度,来确定这个珠子的正常的信号强度。有了标准的信号强度之后。后面测到的信号,都和这四个碱基的信号强度进行对比,如果是一倍强度,我们就知道有一个碱基,如果有2倍的强度,就知道串联了2个相同的碱基,依此类推。
在整个的测序过程中,影响有效数据量的因素有以下这些:

第一, 一张芯片上,在所有的预制孔中,有多少孔是有珠子进入的。ISP density,也就是Ion Sphere™ Particles (ISPs)。比较理想的情况下,ISP density可以达到60~80%之间。这个值一般是由把珠子加到芯片上去的这个过程所决定的,加载得越好,则有珠子的孔数越多,没珠子的孔越少

第二, 是珠子上是否长了文库DNA链。这个指标,是由磁珠纯化的过程来决定的。纯化得越好,则有文库DNA的珠子越多,没文库DNA的珠子越少

第三, 是单克隆的珠子、和多克隆的珠子的比例。所谓单克隆的珠子,就是在一个珠子上只长了一种DNA分子。而多克隆的珠子,是指一个珠子上长了2种或者2种以上的DNA分子。

-
在Ion Torrent测序过程当中,只有单克隆的珠子才可能产生有用的数据。而多克隆的珠子所产生的数据是乱的,是没有用的。
-
产生单克隆的珠子,还是多克隆珠子,是在油包水PCR过程当中,一个水滴当中包含了几个DNA文库分子来决定的。如果一液滴中,一开始只包含了一个文库分子,做出来就是单克隆的珠子。如果一个液滴当中包括了2个、或者2个以上的文库分子,做出来,就会是多克隆的珠子。
-
那么产生多少个多克隆的珠子,又产生多少个单克隆的珠子,它是一个统计的过程。是符合泊松分布的。
-
目前,理想情况下,大概可以达到70~80%左右的珠子是单克隆的珠子。
第四, 是珠子上长的是否是有用的样本序列
-
所建文库当中多多少少会含有一部分的引物二聚体。引物二聚体的序列,是无用的序列
-
测到的序列当中,有一部分的序列质量低于可接受的水平,这是会被去掉的
-
还有,在测序过程当中,一般会加1%的阳性对照珠子,这些对照珠子是进行质量控制的,但是这些阳性对照珠子上所测到的序列,也是无用的序列。
第五, 剩下,是有用的样本序列。
Ion Torrent 测序平台的特点
到目前为止,Thermo Lif公司在Ion Torrent平台上,推出了两款测序仪,小的叫PGM测序仪(Personal Genome Machine的意思),大的叫Proton测序仪。
Ion Torrent平台的主要测序优势,是可以从很少量的起始DNA来进行测序。一般情况下,5~10个ng的DNA就足够进行一次质量良好的测序了。
这个优势,是基于Thermo Life公司推出了一系列基于多重PCR的建库方案。
AmpliSeq是Ion Torrent平台上很好用的一个建库方案。它的核心,就是通过多重PCR的方法,一次从样本中把要测序的多个DNA片段给扩增出来,然后转化成文库进行测序。
ThermoLife公司基于其多年做PCR的经验,推出了这个AmpliSeq平台。在AmpliSeq平台上,一方面推出了许多成熟的测序Panel(,另外客户可以在AmpliSeq平台上,自行订制适合自己的AmpliSeq Panel。
所谓Panel就是一系列针客户感兴趣的基因片段,所做的扩增引物、或者杂交探针的组合。
有兴趣的同学,可以登录“www.ampliseq.com”这个网站,来寻求适合自己的测序服务。
Ion Torrent测序平台,一直有一个Homopolymer的问题。所谓Homopolymer问题,就是测序仪在测到一连串相同的碱基时,就读不准到底有几个碱基。比如说,有一串5个A,测序仪在读到这里的时侯,读到一个强的A信号,但是仪器在判断,这个信号到底是5个A,还是4个A、或者是6个A的时侯,有可能会判断错误。这就是我们所说的Homopolymer错误。

Thermo Life公司最近推出的了Hi-Q酶,这个Hi-Q酶的特点就是聚合反应非常快,也就是它所产生的PH值的变化的峰,更高、更尖、更利于判读。这在相当大程度上提高了Ion Torrent测序仪读Homopolymer区段时的准确性。
油包水PCR反应是一个对操作很敏感的实验步骤。为了提高实验结果的一致性,也为了减少人工消耗,Thermo Life公司还在Ion Torrent平台上推出半自动的油包水PCR反应仪:“One Touch”,和全自动的油包水PCR反应仪:“Ion Chef”
Ion Torrent平台的测序仪,还有一个优点,就是它的测序速度比较快。一般上机测序时间,在3~5个小时左右。
以上是本期视频的全部内容。
5. X10人全基因组测序
本期节目,我们主要给大家介绍:人全基因组测序。前半部分,我们会介绍一下Illumina 公司最新的HiSeq X10,这个专门用于人全基因组测的测序仪。那么后半部分呐,我们会给大家介绍一下人全基因组测序,将给出哪些有用的生物信息。
Illumina HiSeq X Ten
那么我们先来说一说,Illumina的HiSeq X10这个机器。这台仪器是Illuminad 2014年年头上,Illumina公司推出的一个重磅机器。它最吸引人们关注的特点,是它把测一个人全基因组90G数据量的成本,一下子降到了一千美元以下。
这台机器有以下的特点:
1、 首先,它测序的速度非常快,上它现在跑一圈、2张芯片,3天就跑完了。比之前的HiSeq 2000跑机器的时间—11,整整快出了2倍以上;
2、 第2点,就是它每张芯片的数据产量非常大,它的每张芯片的数据产量,可以达到0.9~1个T(10的12次方),甚至更多一些的数据产量。相比于HiSeq 2000每张芯片产0.3个T数据产量,又多出了2倍以上;
3、 第3点,是它的读长又增加了,它的读长从HiSeq 2000的双端(各)100个碱基,增加到现在的双端(各)150个碱基;
4、 最最重要的一点,还是测序成本的大幅下降。Illumina把HiSeq X10平均到每单位(G)数据量的、测序试剂的成本做了大幅的下调。根据Illumina公司自己公布的价格来看,测一个人全基因组90G的数据,它所需要的建库、和测序的试剂成本是797个美金。那么再加上一些人工、仪器折旧的直接成本,这样测一个人全基因90G的直接成本,已经降到1000美元以下。这就是它最大的吸引人的特点。
X10 的技术创新点
X10这台测序仪,之所以可以把测一个全基因组的成本控制在1000美金以下,是因为采用了一系列的技术改进。

NanoWell技术
那么,第一项改进就是用了Nano Well技术。
Nano Well技术就是把原来测序芯片上平铺那2种引物,做成小孔。这些小孔整齐地呈蜂窝状排列,只在小孔的里面,才有长Cluster的引物,小孔之间的那些表面,是没有这些引物的。
这样的改进,所带来的好处:
第一个就是Cluster可以长得更密。
因为现在只有在小孔中有长cluster的引物,所以cluster也只能长在小孔中,原来这些cluster是在一个整个的表面上随机长的,那么必然会有些地方长得密,有些地方长得稀。那么那些长得稀的地方,它的表面实际上就是被浪费掉了。现在把所有的孔都排列起来之后,所有的表面都被充分地利用起来。根据Illumina的官方数据,X10的芯片上,它的表面的cluster密度可以达到一个平方毫米里面有120万个点,那么相对于HiSeq 2500每平方毫米60万个光点来说,那么它(X10的cluster密度)整整高出了一倍。这是第一个好处。
第二个好处,就是它更有利于扫描仪对cluster的判读。
那么原来的这些Cluster,长得就象天上的星星,是随机长的。这样扫描仪要判断Cluster,就需要有一个横向的对比,也就是一个cycle(循环)里面,4个光、4幅照片之间的对比。还要做纵向的对比,也就是不同cycle(循环)之间,要进行光点的对比。那么这样,对扫描仪的扫描精度高求很高,同时,又对服务器的计算,又提出了很高的通量要求。现在改成所有的格子,它的位置都是事先知道的,都是排整齐的。那么,对于光学扫描仪的扫描精确度的要求,就降低了。然后,它(扫描仪)的扫描速度,也可以提上去。
扫描完之后,对光点的空间位置的判断也变得容易,那么计算,也更加高效率了。

RPA技术


那么X10上的第2项改进,就是它的簇生成的技术,从原来的桥式PCR技术,改到了新的RPA技术。
那么RPA技术,是recombinant polymerase amplification的首字母缩写。
那么RPA技术,它是一种模拟自然条件下的DNA扩增技术。
我们知道PCR是用热来解链DNA的。那么RPA技术呐,不再靠热来解链DNA。它是用解链酶、和DNA单链稳定蛋白来把DNA的双链打开,然后再利用聚合酶来合成新DNA链。
RPA技术带来的第一个好处是,就是直接提升了小孔的利用效率。
如果还是用桥式PCR,那么在理想情况下,所有的孔当中,大概有约1/3的孔是单克隆的;还有1/3的孔,会是空克隆。还有1/3的孔,是多克隆。
那么空的孔、和多克隆的孔,都是没用的。
那么空的孔,因为当中没有数据。而多克隆的孔呐,当中的数据是乱的。所以,这两者都是没用的。只有单克隆的孔,是有用的。
那么因为限于泊松分布的这个原理,所以,桥式PCR的方法来做克隆,一般只有1/3的孔是有效的。
那么现在采用了RPA技术之后,它得到这样一个效果:就是这个测序的模扳,和小孔当中的引物的结合,是一个比较慢的过程。但是它一旦结合了之后,它就马上发生扩增。那么经过扩增之后,马上就把一个小孔给占满。
这样,这个小孔即使后面再有新的模板来,那么也无法发生新的扩增。因为已经被占满了。那么,这个有点象地铁上的抢座位,谁先抢到了,谁就坐。那么后来的人,就没位子了,只能站着。
那么,这个呢大大提高了小孔的利用效率。那么根据Illumina官方的数据,它的孔的利用率从原来的1/3,提高到了现在的60%、或者更高。
那么,RPA技术还带来了另外一项好处,就是它对于加入的模板的浓度(变化)的耐受性,更好了。那么现在它对加入的模板浓度(变化)的耐受性,扩大到了20%,而以前,一般只有5~10%的耐受范围。
更快的光学扫描
X10的第3项技术改进,就是用了更快的光学扫描仪。那么它的这个光学扫描速度,是原来HiSeq 2000的光学扫描仪的扫描速度的6倍。
对修饰dNTP更适应的酶
X10的第4项重要改进,是用了新版的酶。


因为在Illumina的测序过程中,用的dNTP并不是天然的dNTP,而是经过了修饰的dNTP。那么这个dNTP,主要有2个修饰。
第1个(修饰),在3’端的羟基上,它带了一个叠氮基团,这个基团,是可逆终止的化学基团,那么这是第1个修饰。
那么第2个(修饰),是在它的碱基上连出去一个长柄,长柄上,再接一个荧光基团。那么就是靠这个荧光基才的颜色,来判断这是哪个碱基。因为接这个荧光基团,所以它的碱基也不是个天然碱基,这是在dNTP上的修饰。
第2个,在边合成边测序过程中生成的这根新的DNA链,它不是个天然的链。因为每个循环结束之后,我们要用化学方法把那个荧光基团给切掉,但是这个切断的位置,并不是齐根的。也就是说每次切断之后,实际上,在这个碱基上还会延出去一个柄。所以,这根新合成的这个DNA链,并不是一个天然的DNA链。
那么这样呢,我们所用的这个聚合酶,它所面对的聚合的环境、和条件并不是天然的条件,它面对的是修饰过的dNTP,同时这个DNA链,也是一个带柄的这样一个DNA链。

那么,每次Illumina的测序技术有大的改进,都会要求这个酶有大的、新的突破,它所有的突破,就是说这个酶能够更好地耐受dNTP的修饰,和耐受这个DNA上的那个多出来的柄。同时,这个酶还要有很高的聚合活性。
那么我们看到,这次,Illumina在HiSeq X10上,它整个的化学速度提高了4倍,那么这个核心的提升,就是这个酶的活性更高。同时,这个酶对这些修饰和变化的耐受性更好了。这就是它的第4个重要改进。
在综合了上述几项重要的技术改进之后,X10测序仪就呈现出了今天我们所看到的这样强大的测序能力。
X10所测出来人全基因组数据能为我们带来的生物信息
在说完了X10的新技术、和强大功能之后,我们再来说一下,它所测出来的数据,能给我们提供什么样的生物信息。
SNP
首先,我们就要说一下,它所提供的SNP信息。
所谓的SNP,就是单核苷酸多态性,也就是Single Nucleotide Polymorphism。这个SNP指的是一个碱基的替换,比如:A变成T,或者G变成了T。这些,都叫SNP。
根据Macrogen公司所发表的30倍测序深度的X10的测序结果,在整个基因组中,98%的区域可以10倍以上的覆盖深度。
有了10倍以上的覆盖深度以后,来确认SNP信息,就相当可靠了。
同样Macrogen公司宣布的X10的测序结果,它所确认的SNP信息,与用HiSeq 2000所确认的SNP信息,互相的比对确认程度,高达到95%以上。这个,也说明X10的测序结果的可靠性是非常之高的。
那么在一般情况下,一个普通黄种人的基因组,与国际公认的、标准的HG19这个参考基因组序列相比,会有350万个左右的SNP。那么这350万个SNP当中,又有大概2万个是落在外显子上的。
而在这2万个落在外显子的SNP当中,非同义的SNP,有大概9千个。所谓非同义的SNP,也就是说这些SNP是会引起蛋白质的序列变化的。
Indel
X10测序数据所能提供的第2种生物信息,就是小片段的DNA插入、和缺失信息。我们通称称这种小片段的插入、缺失叫“Indel”。Indel是“Insertion”和“Deletion”两个英文词的合并缩写。
我们通常所说的Indel是指小于50个BP以内的微小的插入、和缺失突变。
那么Indel如果一旦落在外显子区域,它是一定会引起蛋白质序列变化的。如果它引起的是移码突变,那么在移码位点之后,所有氨基酸序列就和原来的序列完全不同了。
如果它(基因)还能保持原来的阅读框,也会引起蛋白质中若干个氨基酸的增或者减。
一个普通黄种人的基因组和标准的HG19参考基因组相比,约有50万个Indel。其中落在外显子上的,大概在1千个左右。
SV
X10测序数据所能提供的第3种信息,是基因组结构变异信息。我们通常称之为SV信息,也就是:Structure Variation信息。
它是指染色体的结构变异,常见的染色体结构变异,有以下几种:
1、 染色体内部的位移

2、 染色体之间的位移

3、 大片段的缺失


4、 大片段的插入

5、 大片倍的加倍

6、 大片段的倒位

CNV

X10测序(数据)所提供的第4种信息,是拷贝数变异信息。也就是CNV信息,Copy Number Variation信息。
它是指染色体片段的拷贝数变异。它包括拷贝数增加,也包括拷贝数减少。
实际上,CNV是和结构变异(也就是SV)紧密相关的。
结构变异中的大片段的增加、和大片段的缺失,会直接导致CNV的变化。
人全基因组测序,它对CNV是高度敏感的。这儿是一张图,来告诉我们:在全基因组测序当中,这些大片段的缺失和增加是可以被非常明确地给测出来的。
测序深度
这里,我们要说明一下,我们所说的,测90个G,或者测30倍的深度,对于测germline变异,也就是我们通常所说的“娘胎里带来的变异”,一般来说是够了。
因为一个人的基因,一半来自于爸爸,还有一半来自于妈妈。那么这两半,基本上是各占一半,所以,一个基因2个拷贝。每个拷贝,如果能够被测到5倍或者10倍,实际上做基因分型,是够了。
那么如果要测肿瘤,那是不够的。因为肿瘤的突变,是Somatic突变,也就是我们说的“体细胞突变”。
因为肿瘤细胞是和正常细胞混着长的,那么在肿瘤组织里面,正常细胞往往还占了大多数,肿瘤细胞往往只占一半、甚至更少一点。那么肿瘤细胞中的一个基因的2个拷贝,往往是1个发生了某种突变。所以,这个情况下,这个突变的基因,在整个这一群细胞当中占的比例会低于25%,甚至更低。甚至只有百分之几。
如果,这时候,测序只测30倍,是比较难于测到这些低频突变的。
那么为了要测到这些低频突变,这就要求我们对肿瘤要加大它的测序深度。那么,目前科研人员一般会对实体肿瘤进行50倍到100倍深度的(全基因组)测序。
同时,还会测一下病人血液中白细胞的基因组DNA(一般是测30X,90G)。那么,把这个白细胞的基因DNA作为正常背景,来做对照。然后,把肿瘤测出来的序列,和正常的对照,进行比对。来找出当中的突变。
还有一些科学家,会做另外一个选择,他会对肿瘤和血液白细胞的DNA,都做30X的(人全基因组)测序(目的是得到肿瘤CNV、SV信息),但是,他们会对肿瘤加测一个100X或者200X的外显子测序。
那么有了这些高深度的测序之后,我们才有比较大的把握,来捕捉到肿瘤中的那些低频的基因突变。
关于这4种突变,1、点突变;2、插入、缺失突变;3、结构变异;4、拷贝数变异,与肿瘤的发生有什么关系,大家可以在我的微信公众号【陈巍学基因】中回复“视频4”三个字,就可以看到我之前给大家做的,关于这方面的介绍视频了。
6. illumina 公司的Nano Well 测序技术
(Illumina公司)专利的微流体芯片是新的创新测序技术。极大地增加了数据的产量、和通量。让我们来仔细看一下此项新技术。
数十亿个纳米级的小孔按预先设定好的方式,整齐地排列。保证了DNA簇有固定的大小、和空间位置,使高密度芯片同时又有精确分辨率成为可能。

好处是数据产量大、有更多序列条数、和更快的仪器运行速度。
现在,你可以在更短的时间内处理更多样本。
这个流式芯片是用数十亿个有专利的Nano Well小孔,在玻璃基质上制作出来的。
在流式芯片生产好之后,只有在小孔中,才种有DNA引物。

在生成DNA簇的时候,新的、独家的扩增技术,保证一个小孔中,只有一个DNA模板,会与(小孔中的引物)结合,并形成簇。
当DNA模板与种子引物相结合时,它会立即快速扩增。这种快速的扩增,能防止别的DNA模板在同一个小孔中,形成“多克隆”。


这就保证了,在每一Nano Well小孔中,只形成来自一个模板的单克隆簇,这样做的结果,就是在大部分的小孔中,每个小孔都是来自于一个模板形成的DNA簇,当这种单模板簇形成好之后,流式芯片就可以做测序了。
测序过程是用行业内公认的、领先的、边合成边测序试剂来进行。
我们承诺:开发创新的测序解决方案,让突破性的发现成为可能,并释放基因组的(巨大)力量。

7.第一代DNA测序
本期节目,我们会谈一下:第一代DNA测序。
第一代测序,又称“Sanger”法测序,或者叫“双脱氧法”测序。
这是由美国生物化学家Frederick Sanger先生发明的,Sanger先生也因为此项发明,而获得诺贝尔奖。
ABI公司(现为ThermoFisher的一部分)在Sanger先生的双脱氧法的基础上,进一步开发出,荧光标记的双脱氧法测序试剂盒。也就是分子生物学界鼎鼎大名的BigDye 试剂。

接着,(ABI)再结合毛细管电泳,生产出了“ABI 3730”和“ABI 3500”等非常成功的测序仪。
到目前为止,ABI 3730、ABI 3500等测序仪和BigDye测序试剂,都是业内公认的一代测序的金标准。
今天,我们就以BigDye试剂为主线,来介绍一下,第一代测序方法的原理。
双脱氧法测序的第一个核心技术:就是在用DNA聚合酶合成DNA链的过程当中,掺入双脱氧核苷酸,也就是“ddNTP”,英文叫“Dideoxynucleotides”。
天然的DNA的组成元件是单脱氧核苷酸,也就是dNTP,deoxynucleotides。在其糖基的5’位、和3’位,各有一个羟基。5'位的羟基,连到上游的磷酸基团;3'位的羟基,连到下游的磷酸基团。这样不断重复,就形成了DNA的一条骨架链。
这就象一群人,其中每个人都伸出双手,左、右手,都各拉住一个伙伴,这样,就形成了一条长长的链。
Sanger的方法,就是用化学合成的办法合成出3’位没有羟基的核苷酸,也就是双脱氧核苷酸(ddNTP)。它比单脱氧核苷酸少了一个3’位的羟基,只保留了5'位的羟基。
它就象一个只有左手、但没有右手的独臂人,在聚合反应当中,它可以被聚合酶结合到DNA链当中去。因为它缺了3'位的羟基,所以它就没有办法和下一个dNTP结合了。DNA链的聚合反应也就此终止,不再往下延伸了。


这样,在DNA链聚合过程当中,通过掺有ddNTP的dNTP进行聚合反应,得到一系列不同长短的DNA片段。每个片段的3’位末端,都是一个双脱氧的核苷酸残基。并且这个核苷酸的残基,是与模板上对应位置的碱基互补的。

接下来,我们来说BigDye试剂的创新点。
它在双脱氧核苷酸(ddNTP)的基础上,再在碱基上,加上荧光发光基团,并且,A、G、C、T四种碱基各标一种颜色的发光基团。

有了不同颜色的荧光发光基团做标签,在最后的识别过程当中,就可以方便地通过颜色,就识别出这末端的双脱氧核苷酸碱基是哪种碱基。
在实际的测序中,先在反应体系当中,加入要测序的DNA模板。一般是经过纯化的质粒,或者经过纯化好的PCR扩增片段。
再加入与测序起始位置已知序列相互补的测序引物DNA,也就是primer。测序Primer在这里起的作用,是与模板的特定序列位置相结合,引导聚合反应发生。并且,它还可以确保:DNA的聚合反应,是从已知的、确定的起点开始。
然后,加入BigDye试剂,进行反应。
BigDye试剂当中,包括了刚才我们所说的“四种荧光标记的双脱氧核苷酸”、dNTP、和DNA聚合酶。另外,还包含了镁离子、PH缓冲液等。
反应过程当中,聚合酶从Primer处开始进行聚合反应。荧光标记的双脱氧核苷酸和天然dNTP,遵照碱基互补的原则,延着模板,一个、一个地被聚合到新合成的DNA链上去。
每聚合一个新的碱基,都有2种可能。
第一种可能,是结合进了一个正常的、与模板互补的dNTP。这时候,聚合反应就可以续继进行下去。

另外一种可能,是结合进一个与模板互补,但是双脱氧的、荧光标记的ddNTP。当DNA链中被结合进了一个ddNTP的时候,链的延伸就被终止。同时BigDye的荧光基团也就被加到这个DNA链的3'位末端。并且这个荧光基团的颜色,与模板对应位置的碱基种类,有对应的关系。
整个反应过程当中,产生了一系列、长长短短的、分别带有荧光标签的DNA片段混合物。
接着,这些DNA片段的混合物,经过一个简单的纯化,去掉游离的荧光ddNTP单核苷酸,留下有一定长度的DNA片段,就可以上机测序了。

上机测序过程当中,先在一根长长的、中空的玻璃毛细管当中,注入丙烯酰胺溶液。接着用紫外光照射丙烯酰胺溶液,丙烯酰胺在紫外线的电离作用下,发生聚合反应,变成聚丙烯酰胺凝胶。
在电场条件下,聚丙烯酰胺凝胶,对于在其中电泳的核酸,有分离作用。短的片段,在聚丙烯酰胺凝胶当中电泳得快;长的DNA片段,则电泳得慢。
然后,把DNA片段混合物,加到有聚丙烯酰胺凝胶的毛细管的一端。在毛细管的两端,加上高电压。DNA片段就在电场的作用下,从负极向正极电泳。
在毛细管的正极的末端,用激光进行照射,并用分光的光学传感器把不同颜色的荧光强度经记录下来。
每个DNA片段,在通过激光的扫描点时,它上面带有的荧光基团就会发出特定颜色的荧光。


因为在之前的聚合反应过程当中,聚合反应的起点都是从特定的引物位置开始的,所以越先电泳到达激光扫描点的DNA片段,就是越短的片段。它的聚合终止位置,离聚合的起始位置就越近。它所产生的荧光颜色,就反映了它3’端末端的那个碱基是A、C、G、T当中的哪一种。
那么反之,越慢电泳到达激光扫描点的DNA片段,就是越长的片段。它的终止位点,就离引物的起始位置越远。
然后,我们就得到了这样一种有四种颜色的图。

图的横轴是电泳的时间;纵轴是荧光的强度;4种颜色,则对应了4种碱基。
那么横轴既可以看作是电泳的时间,也可以看作是碱基的先后次序。

延着横轴,我们可以根据峰的颜色,判断出,依次是哪种碱基。
峰越高、越尖,与别的峰的交错越少,则这个碱基判读准确性越好。
上述,就是ABI公司BigDye测序方法的基本原理。
目前,用ABI 3500测序仪,一般可以测到850个碱基(长度),或者更长的片段的碱基序列。用ABI 3730(测序仪),一般可以测到700个碱基(长度),或者更长片段的序列。
以上是本期节目的全部内容。谢谢您的收看

 浙公网安备 33010602011771号
浙公网安备 33010602011771号