

拓端tecdat|R语言绘制圈图、环形热图可视化基因组实战:展示基因数据比较
原文链接:http://tecdat.cn/?p=23891
原文出处:拓端数据部落公众号
可以使用环状图形展示基因数据比较。可以添加多种图展信息,如热图、散点图等。
本文目标:
- 可视化基因组数据
制作环形热图
环形热图很漂亮。可以通过R来实现环形热图。
首先,让我们生成一个随机矩阵,并将其随机分成五组。
-
-
mat1 = rbind(cbind(matrix(rnorm(50*5, mean = 1), nr = 50),
-
matrix(rnorm(50*5, mean = -1), nr = 50)),
-
cbind(matrix(rnorm(50*5, mean = -1), nr = 50),
-
matrix(rnorm(50*5, mean = 1), nr = 50))
-
)

下面的图是热图的正常布局。
Heatmap(mat1, row_split = split)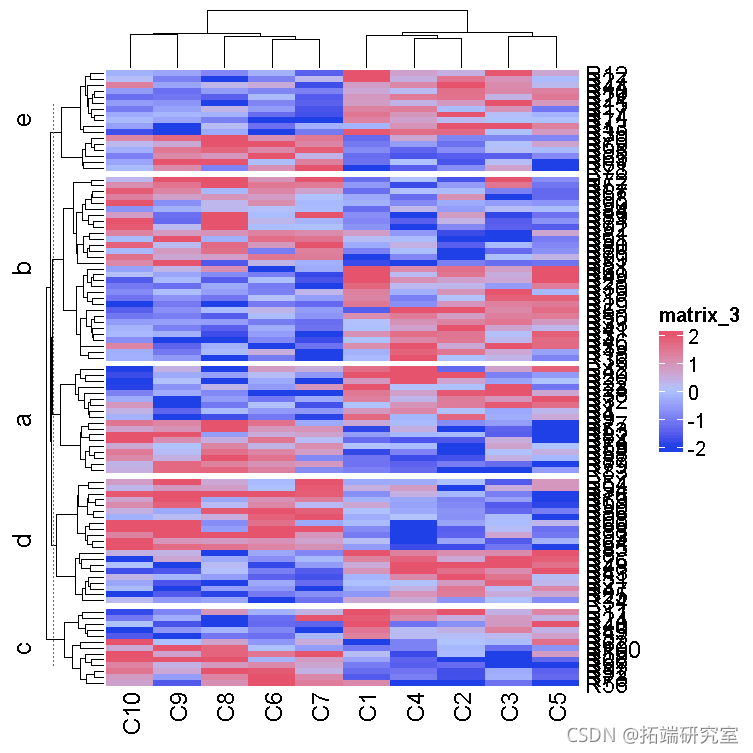
在接下来的章节中,我将演示如何将其可视化。
输入数据
heatmap()的输入应该是一个矩阵(或者一个将被转换为单列矩阵的向量)。如果矩阵被分割成组,必须用split参数指定一个分类变量。注意spilt的值应该是一个字符向量或一个因子。如果它是一个数字向量,它将被转换为字符。
颜色是矩阵中数值的重要美学映射。用户必须用用户定义的颜色模式指定col参数。如果矩阵是连续数字,如果矩阵是字符,col的值应该是一个命名的颜色向量。
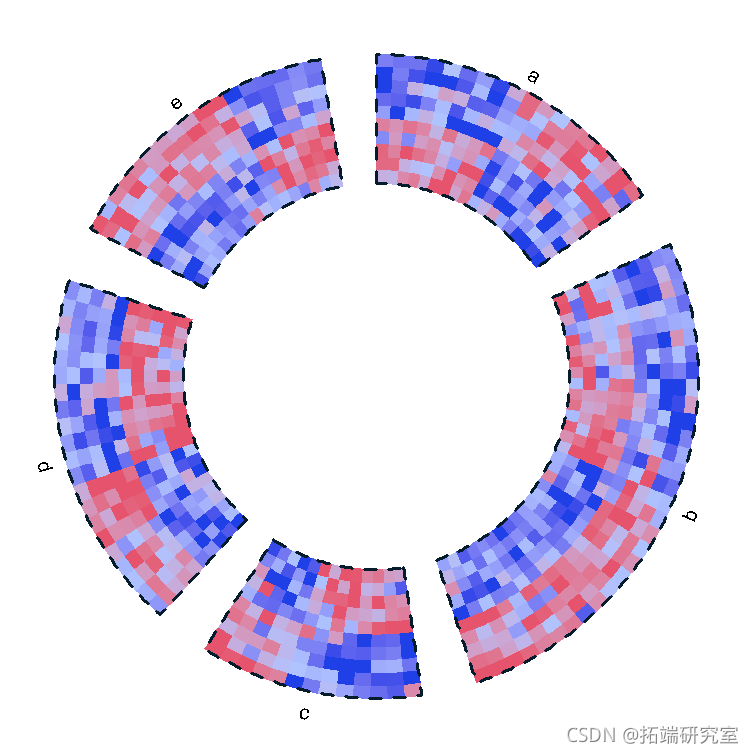
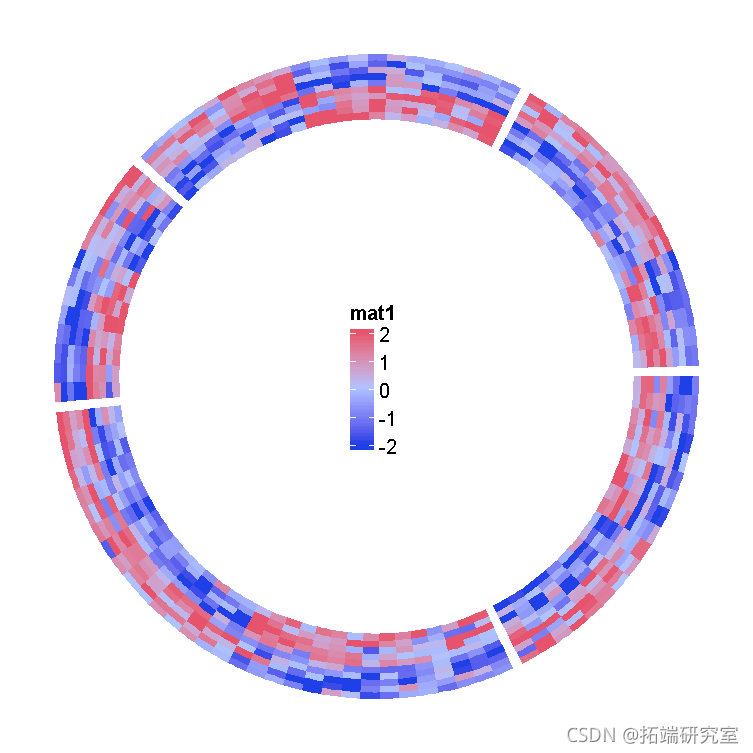
下面的图是之前热图的圆形版本。请注意,矩阵的行沿圆形方向分布,矩阵的列沿径向方向分布。在下面的图中,圆形被分成五个部分,每个部分对应一个行组。
heatmap(mat1col_fun1)
有一件事非常重要,那就是在创建圆形热图之后,你必须完全删除布局。
如果没有指定split,就只有一个大的扇区包含完整的热图。

环形布局
与生成的其他圆形图类似,环形布局可以在制作图之前由par()控制。
热图轨道的参数可以在circos()函数中控制,如track.height(轨道的高度)和bg.border(轨道的边界)。
在下面的例子中,通过设置show.sector.labels参数,增加了扇区的标签。扇区的顺序是c("a", "b", "c", "d", "e"),按时钟方向排列。你可以在下面的图中看到,a扇区从 θ=90∘θ=90∘开始。
-
heatmap(
-
bg.border )
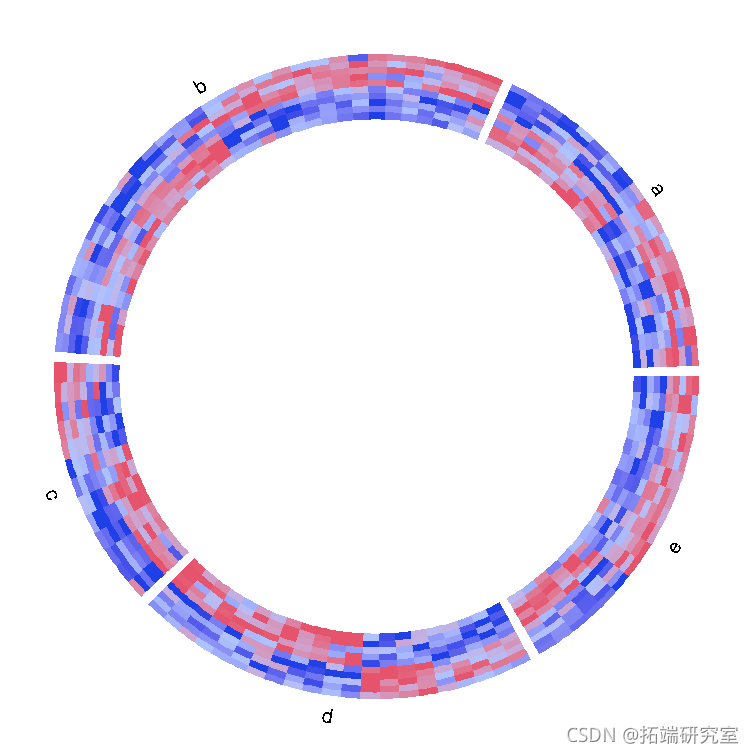
如果split参数的值是一个因子,那么因子水平的顺序控制热图的顺序。如果split是一个简单的向量,热图的顺序是unique(split)。
-
# 注意,因为在前一个图中调用了 circos.clear() 。
-
# 现在布局从theta = 0开始(第一个扇区是'e')。
-
heatmap( levels = c("e", "d", "c", "b", "a))
树状图和行名
默认情况下,数字矩阵是按行聚类的,因此,有聚类产生的树状图。side参数控制树状图相对于热图轨道的位置。注意,树枝图是在一个分离的轨道上。
heatmap(dend.side = "inside")
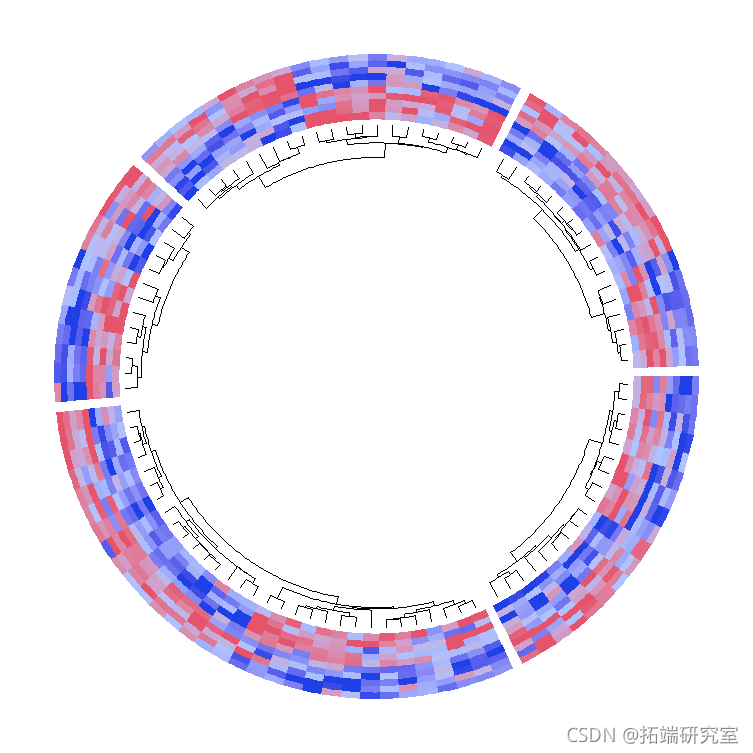
树状图的高度是由dend.track.height参数控制的。
矩阵的行名可以通过设置rownames.side参数来绘制。行名也会被绘制在一个分离的轨道中。
heatmap(rownames.side = "inside")


矩阵的行名和树状图可以同时绘制。当然,它们不能在热图轨道的同一侧。
-
dend.side = "inside",
-
rownames.side = "outside"

行名的图形参数可以设置为标量或向量,长度与矩阵中的行数相同。
heatmap(col = col_fun1, rownames.side = "outside")
树状图的图形参数可以通过回调函数直接渲染树状图来设置,这一点将在后面演示。
聚类
默认情况下,数字矩阵是按行聚类的。 cluster参数可以设置为FALSE来关闭聚类。
当然,当cluster被设置为FALSE时,即使dend.side被设置,也不会绘制树状图。
聚类方法和距离方法由clustering.method和distance.method参数控制。
请注意heatmap()不直接支持对矩阵列的聚类。 你应该在使用heatmap()之前应用列的重新排序,例如。
hclust(dist(t(mat1)))$order
对树状图的回调
聚类产生树状图。回调函数可以在每个树状图生成后应用于相应的类。回调函数可以编辑树状图,例如:1.重新排列树状图,或者2.给树状图着色。
在circos.heatmap()中,一个用户定义的函数应该被设置为callback参数。该用户定义的函数应该有三个参数。
dend: 当前扇区的树状图。m: 与当前扇区相对应的子矩阵。si: 当前扇区的扇区索引(或扇区名称)。
默认的回调函数定义如下,它通过对矩阵行均值加权来重新排列树状图。
reorder(dend, rowMeans(m))下面的例子通过dendsort()对每个扇区的树状图重新排序。
-
heatmap( col = col_fun1, dend.side = "inside",
-
dendsort(dend)
-
}

我们可以使用color()来渲染树状图的边缘。 例如,为五个区的树枝图分配不同的颜色。这里,树枝图轨道的高度由height参数增加。
-
-
den = function(dend, m, si) {
-
# 当k = 1时,它为整个树状图渲染一种相同的颜色
-
color_branches(dend, k = 1, col = dend_col[si])
-
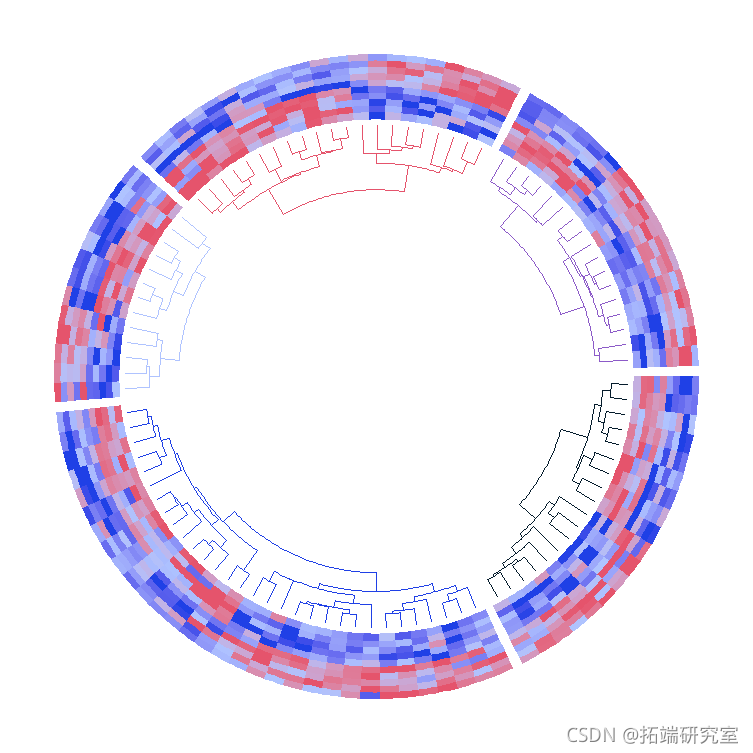
或者如果矩阵没有被分割,我们可以给子树状图分配不同的颜色。
-
-
color_branches(dend, k = 4, col = 2:5)
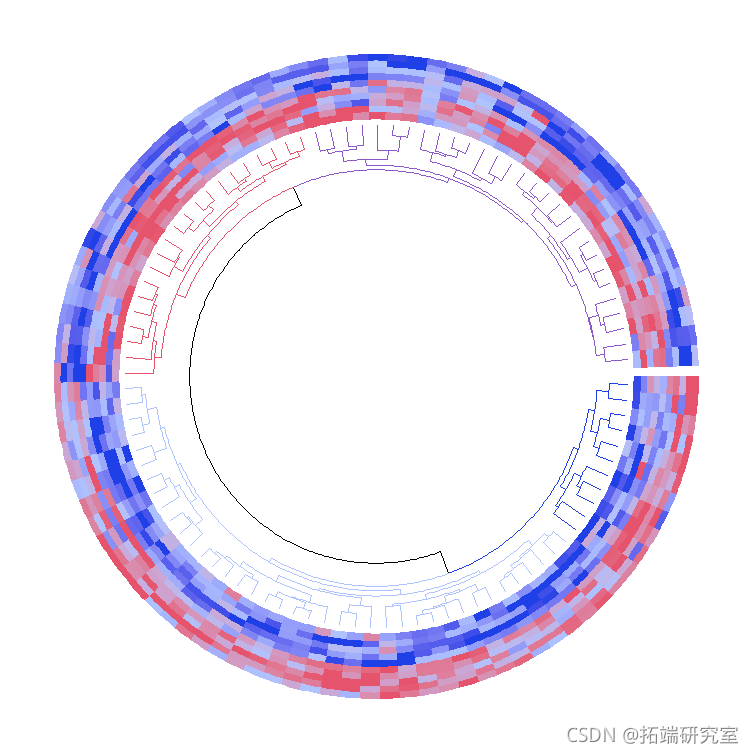
多个热图轨迹
如果你制作的环状图只包含一个热图轨迹,使用heatmap()是非常简单的。如果你制作一个包含多个轨道的更复杂的环状图,你应该了解关于heatmap()的更多细节。
heatmap()的第一次调用实际上是初始化布局,即应用聚类和拆分矩阵。树状图和分割变量是内部存储的。这就是为什么你应该明确地调用clear()来删除所有的内部变量,这样可以确保当你制作一个新的圆形热图时,heatmap()的第一次调用是在一个新的环境中。
heatmap()的第一次调用决定了所有轨道的行顺序(循环方向的顺序),因此,接下来的轨道中的矩阵共享与第一个轨道中相同的行顺序。另外,后面轨道中的矩阵也会根据第一个heatmap轨道中的分割情况进行分割。
如果在第一个热图轨道中没有应用聚类,则使用行的自然排序(即c(1,2,...,n))。
-
-
mat1[sample(100, 100), ] # 按行随机排列 mat1
-
heatmap(mat1, split, col_fun1, dend.side = "outside")
-
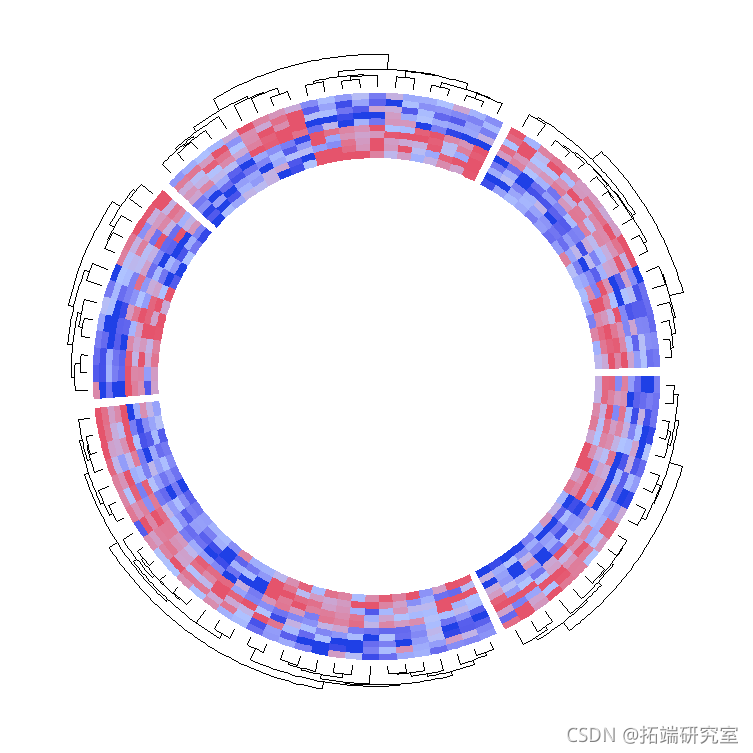
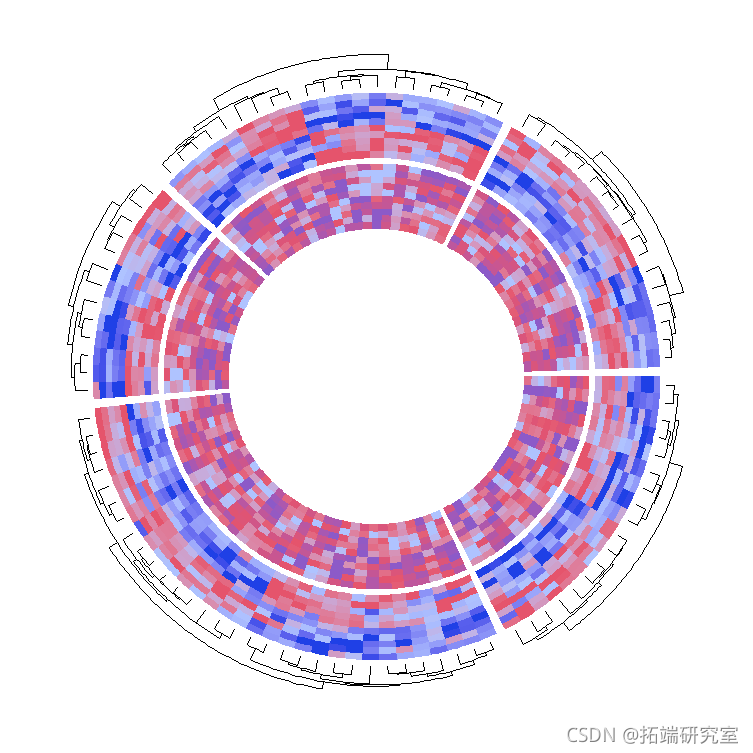
如果我切换两个轨道,你可以看到现在的聚类是由第一个热图轨道控制的,也就是蓝-红热图轨道。
heatmap(mat1, col = col_fun2) 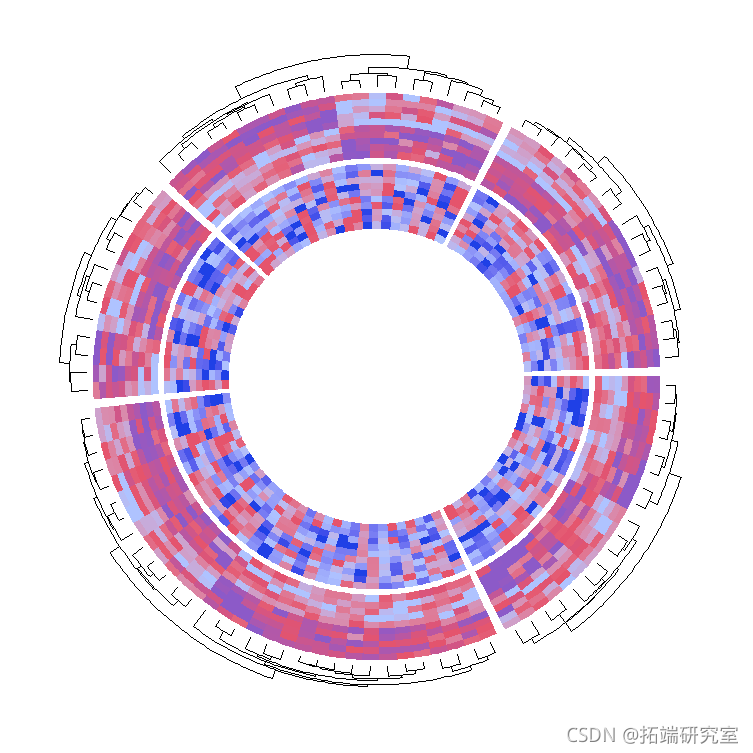
你可能想问,如果我不希望聚类是由第一个轨道决定的,而第二个或第三个轨道呢?解决办法很简单。实际上,初始化可以通过明确调用initialize()函数来手动完成。
在initialize()中,你指定你想应用聚类的任何矩阵以及分割变量,然后,下面的heatmap()调用都共享这个布局。
在下面的例子中,全局布局是由mat1决定的,它在第二个轨道中被可视化。我在第一个轨道中设置了side = "outside",实际上你可以发现树状图实际上是根据第二个轨道中的矩阵生成的。
circos.heatmap.initialize(mat1, split = split)
在下一个例子中,热图布局是由mat1生成的,而两个热图轨道分别只包含五列。
initialize(mat1, split = split)
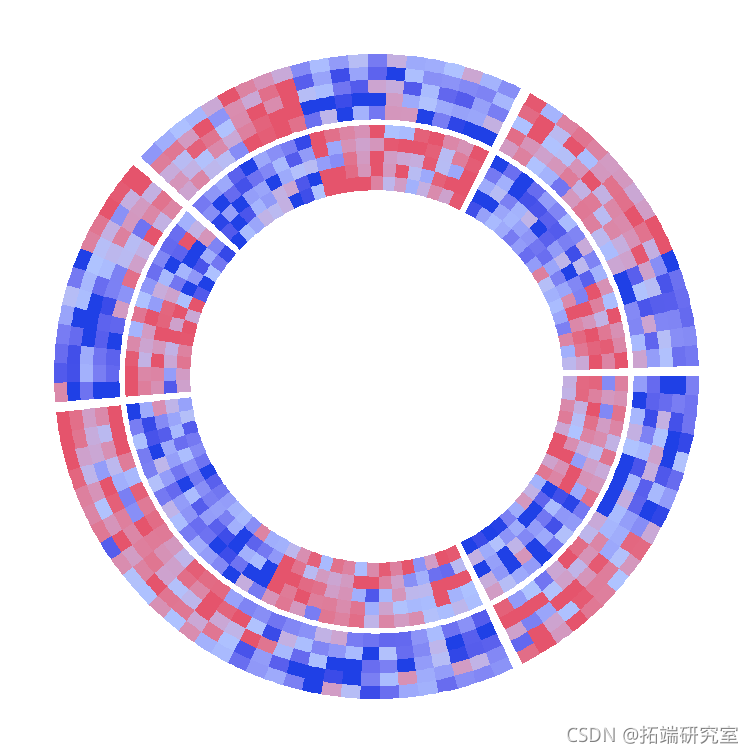
与其他轨道整合
其他非热图轨道整合。在环形布局中,x轴和y轴上的值只是数字索引。假设在一个扇形区域内有nr行和nc列的热图,热图行的绘制间隔为(0,1),c(1,2),...,c(nr-1,nr),热图列也类似。同时,原始矩阵也被重新排序。如果增加更多的轨道,需要考虑所有这些影响,以确保与热图轨道有正确的对应关系。
热图布局完成后,轨道/扇区/单元的额外信息可以通过特殊变量CELL_META来检索。单元/扇区的附加元数据列举如下,它们对于正确对应热图轨道非常重要。
CELL_META$row_dend或简称CELL_META$dend:当前扇区的树状图。如果没有进行聚类,则该值为NULL。CELL_META$row_order或简称CELL_META$order:聚类后当前扇区中子矩阵的行排序。如果没有进行聚类,其值为c(1, 2, ..., )。CELL_META$subset。原始完整矩阵中指数的子集。这些值的排序是递增的。
以下是CELL_META$row_dend、CELL_META$row_order和CELL_META$subset在循环热图例子中的第一个扇区的输出。
-
row_order
-
-
subset
![]()
![]()
在下面的例子中,我添加了一个轨迹,它将mat1中前五列的行平均值可视化。我添加了cell.padding = c(0.02, 0, 0.02, 0),这样最大和最小的点就不会与单元格的上下边界重叠了。
-
track(ylim = range(row_mean{
-
lines(CELL_META$cell.xlim, c(0, 0),
-
points(seq_along(y) - 0.5, y, )
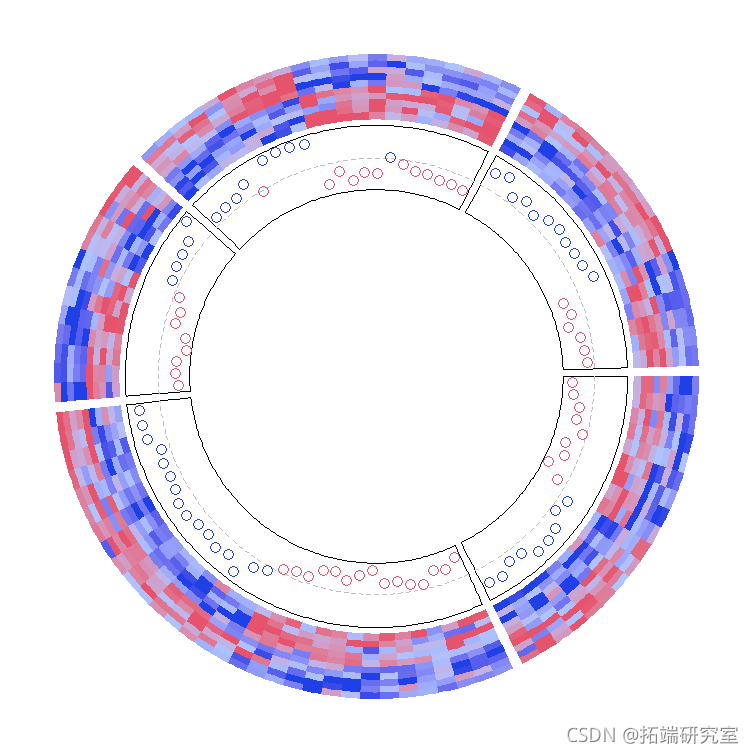
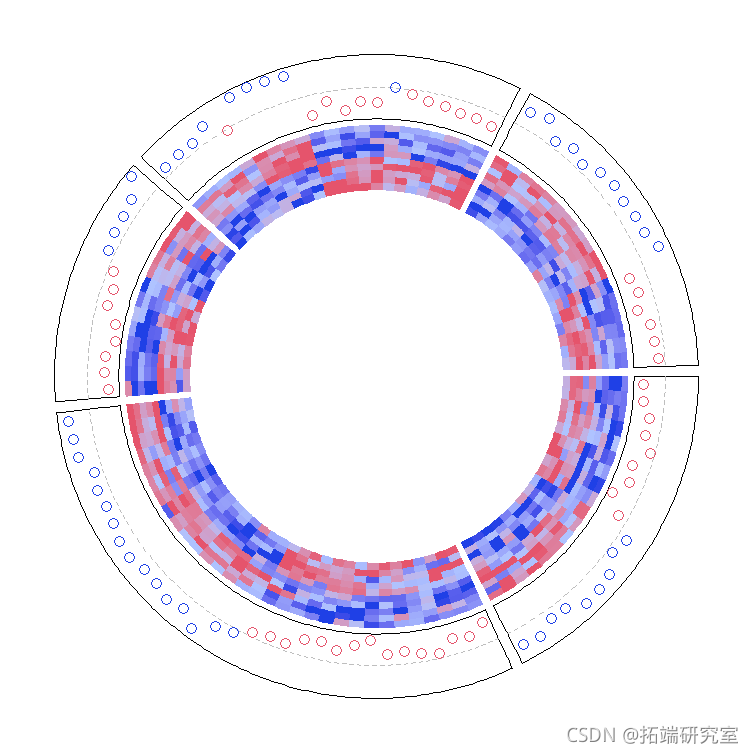
同样地,如果把点数轨道作为第一条轨道,则应事先对布局进行初始化。
-
initialize(mat1, split = split)
-
# 这与前面的例子相同
-
track(ylim = range(row_mean), panel.fun = function(x, y) {
-
circose(y > 0, "red", "blue"))
-
}, cell.padding = c(0.02, 0, 0.02, 0))
-
# 不需要在这里指定 "分割"。
-
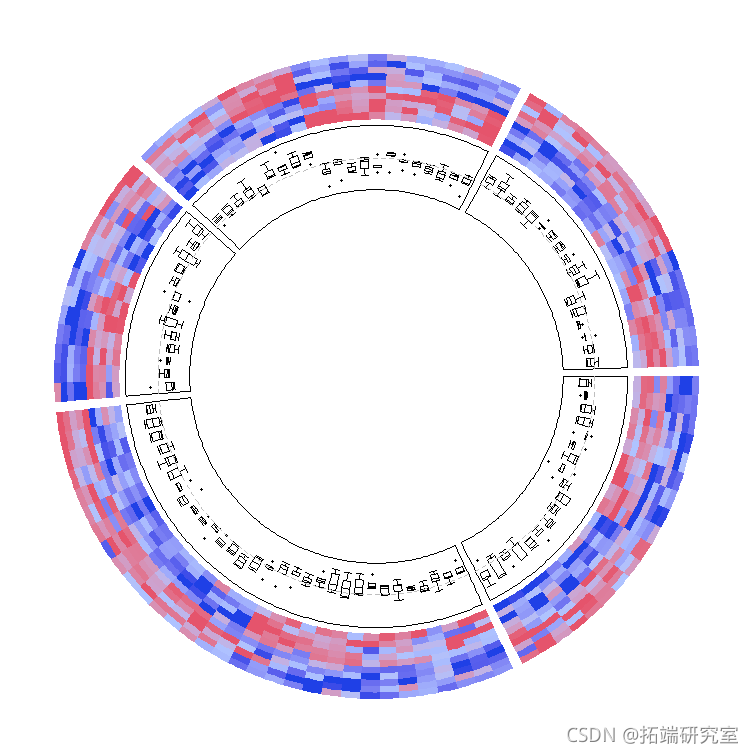
Boxplots被用来对应矩阵的行。
-
circos.heatmap(mat1, split = split, col = col_fun1)
-
circos.track(ylim = range(mat1), panel.fun = function(x, y) {
-
m = mat1[CELL_META$subset, 1:5, drop = FALSE]
-
m = m[CELL_META$row_order, , drop = FALSE]
-
n = nrow(m)
-
# circos.boxplot is applied on matrix columns, so here we transpose it.
-
circos.boxplot(t(m), pos = 1:n - 0.5, pch = 16, cex = 0.3)
-
circos.lines(CELL_META$cell.xlim, c(0, 0), lty = 2, col = "grey")
-
}, cell.padding = c(0.02, 0, 0.02, 0))
添加注释
可以通过设置abels = TRUE来添加区块的标签,然而,这并不提供对标签的任何定制。用户可以通过定义fun函数来定制自己的标签,演示如下。
-
heatmap(col = col_fun1)
-
circos.track
-
adj = c(0.5, 0), niceFacing = TRUE)
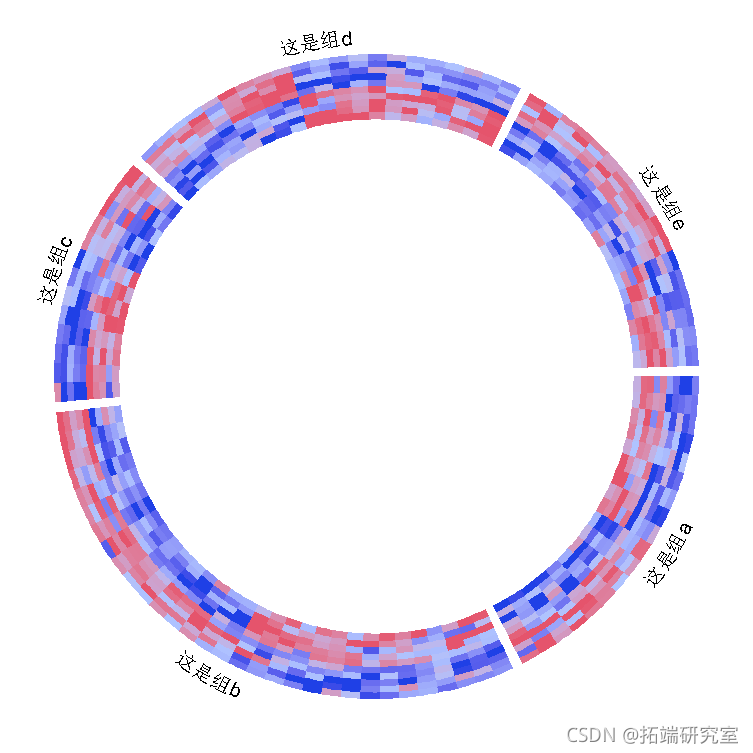
heatmap()不直接支持矩阵的列名,但也可以通过自行定义fun函数轻松添加。在下面的例子中,我通过par()中的after参数在最后一个扇区(第五扇区)后设置了较大的空间(10度,用户通常需要尝试几个值来获得最佳空间),之后我在fun中绘制了最后一个扇区中的列名。
-
par(gap.after = c(2, 2, 2, 2, 10))
-
heatmap(mat1, split , col = col_fun1)
-
track(track.index = 1
-
-
}, bg.border = NA)
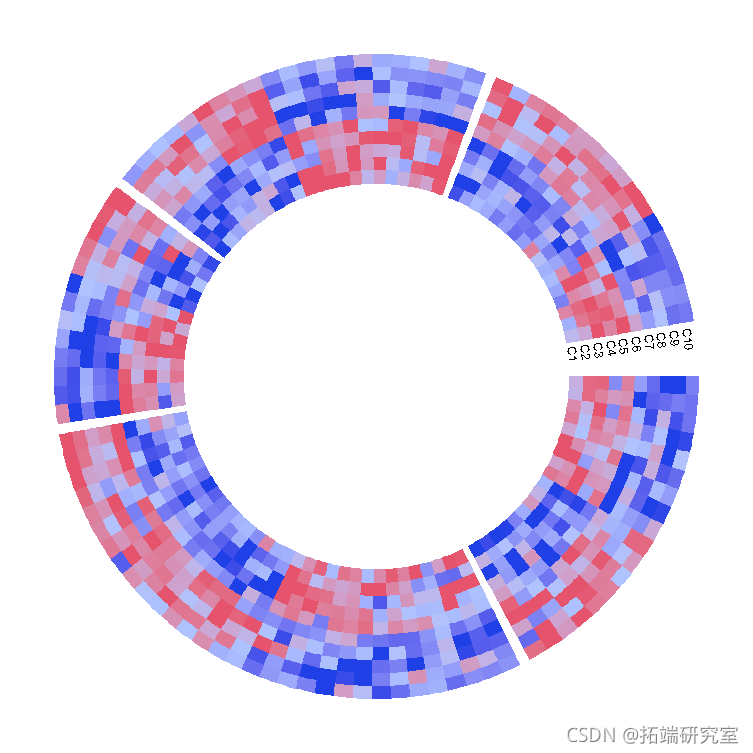
下一个例子添加了矩形和标签来显示矩阵中的两组列。fun里面的代码很简单。convert_x()将x方向上的单位转换为环形坐标系中测量的适当数值。
-
par(gap.after = c(2, 2, 2, 2, 10))
-
heatmap(mat1, split , col = col_fun1)
-
track(track.index = 1
-
circos.text(cell.xlim[2] + convert_x(3, "mm"), 7.5,
-
}, bg.border = NA)
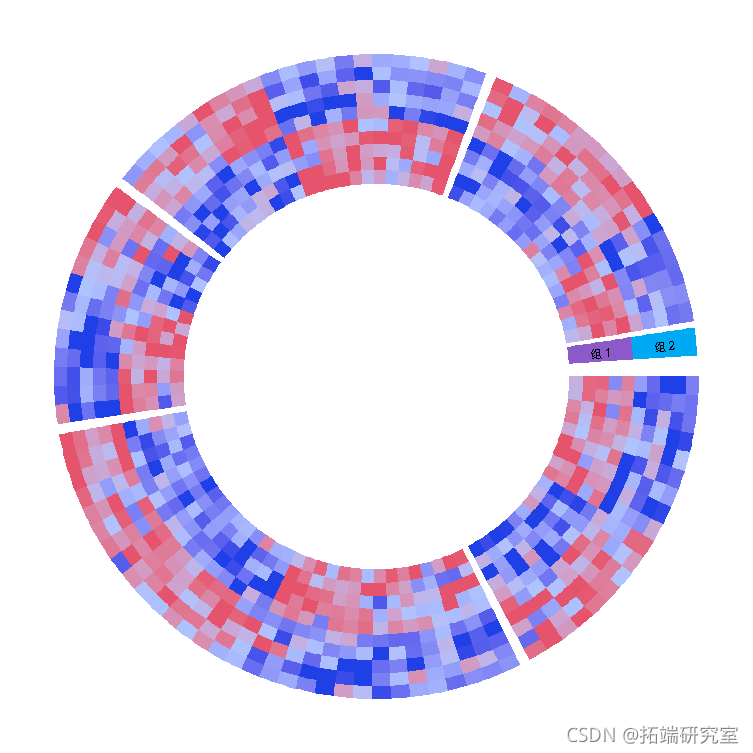
circlize不生成图例,但是图例可以由Legend()函数手动生成并添加到圆形图中。下面是一个添加图例的简单例子。在下一节中,你可以找到一个添加许多图例的更复杂的例子。
-
heatmap(mat1, split = split)
-
clear()
-
grid.draw(lgd)
一个复杂的圆形热图的例子
在本节中,我将演示如何制作复杂的圆形热图。下图是正常布局的热图,现在我将用圆形布局改变它们。
热图直观地显示了DNA甲基化、基因表达和其他基因组水平信息之间的相关性。
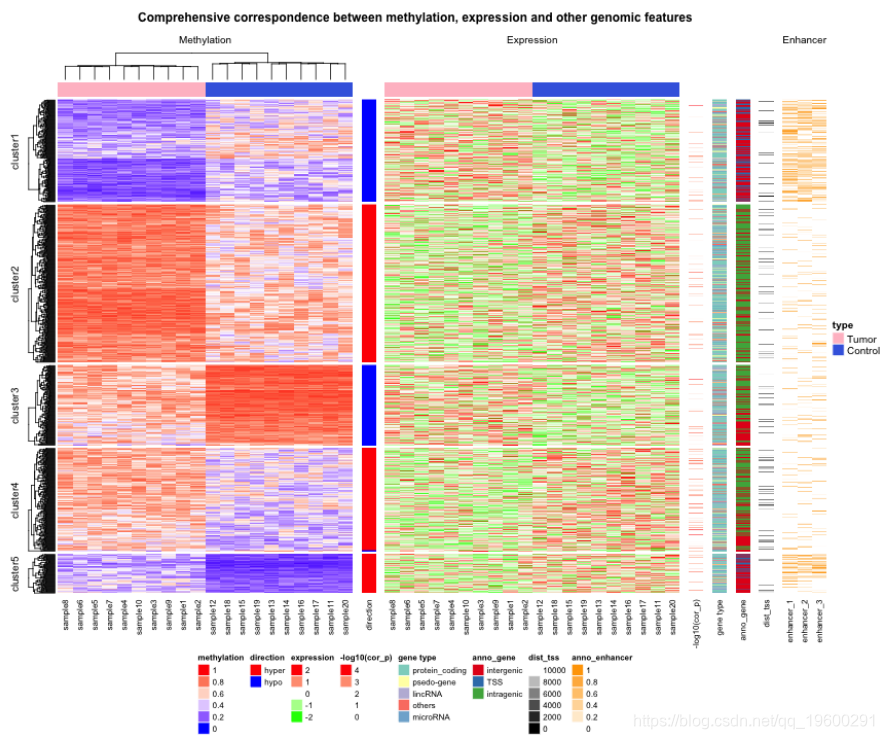
原始热图是用随机数据集生成的。
与原始热图类似,通过对甲基化矩阵(mat_meth)的行进行k-means聚类,将所有热图的行分成5组。
km = kmeans(mat_meth, centers = 5)$cluster现在有以下矩阵/向量需要被可视化为热图。
mat:一个矩阵,其中各行对应不同的甲基化区域(DMRs)。矩阵中的值是每个样本中DMR的平均甲基化水平。expr:一个矩阵,其中的行对应于与DMR相关的基因(即与DMR最近的基因)。矩阵中的值是每个样本中每个基因的表达水平。每个基因在不同样本中的表达量是有比例的。direction:甲基化变化的方向(hyper表示肿瘤样本中甲基化程度较高,hypo表示肿瘤样本中甲基化程度较低)。pvalue:甲基化和相关基因表达之间的相关测试的P值。数值是-log10转换的。type:基因的类型(如蛋白质编码基因或林肯RNAs)。gene:对基因模型的注释(即基因间、基因内或转录起始位置(TSS))。dist:从DMRs到被鉴定基因的TSS的距离。enhancer:每个DMR中与增强器重叠的部分。
在这些变量中,mat_meth、mat_expr、cor_pvalue、dist和anno_enhancer是数字变量,我为它们设置了颜色映射函数。对于其他变量,我设置了命名的颜色向量。
在下面的代码中,我在heatmap()的第一次调用中指定了分裂,这是甲基化热图。轨道的高度是手动调整的。
-
col_meth = colorRamp2(c(0, 0.5, 1), c("blue", "white", "red"))
-
heatmap(mat, split col_meth, track.height = 0.12)
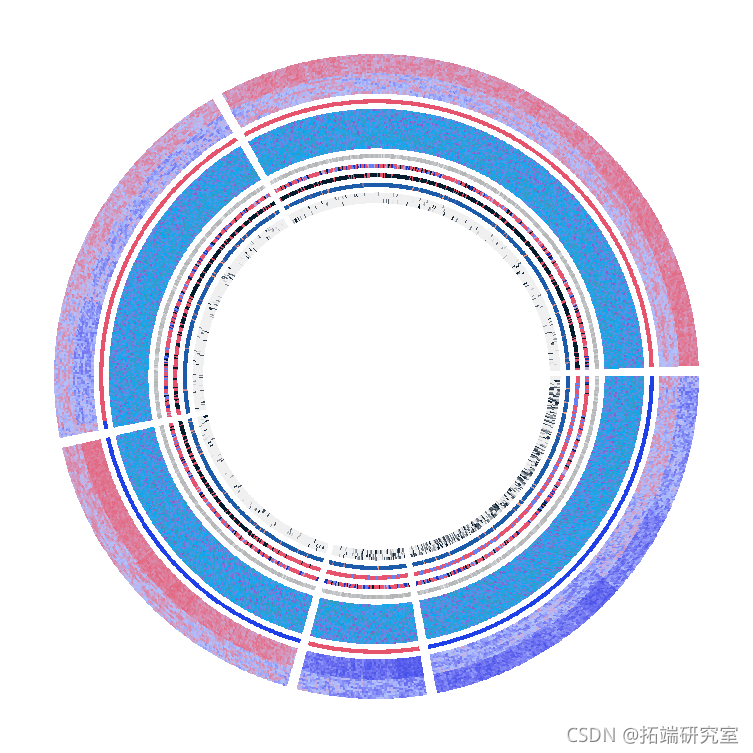
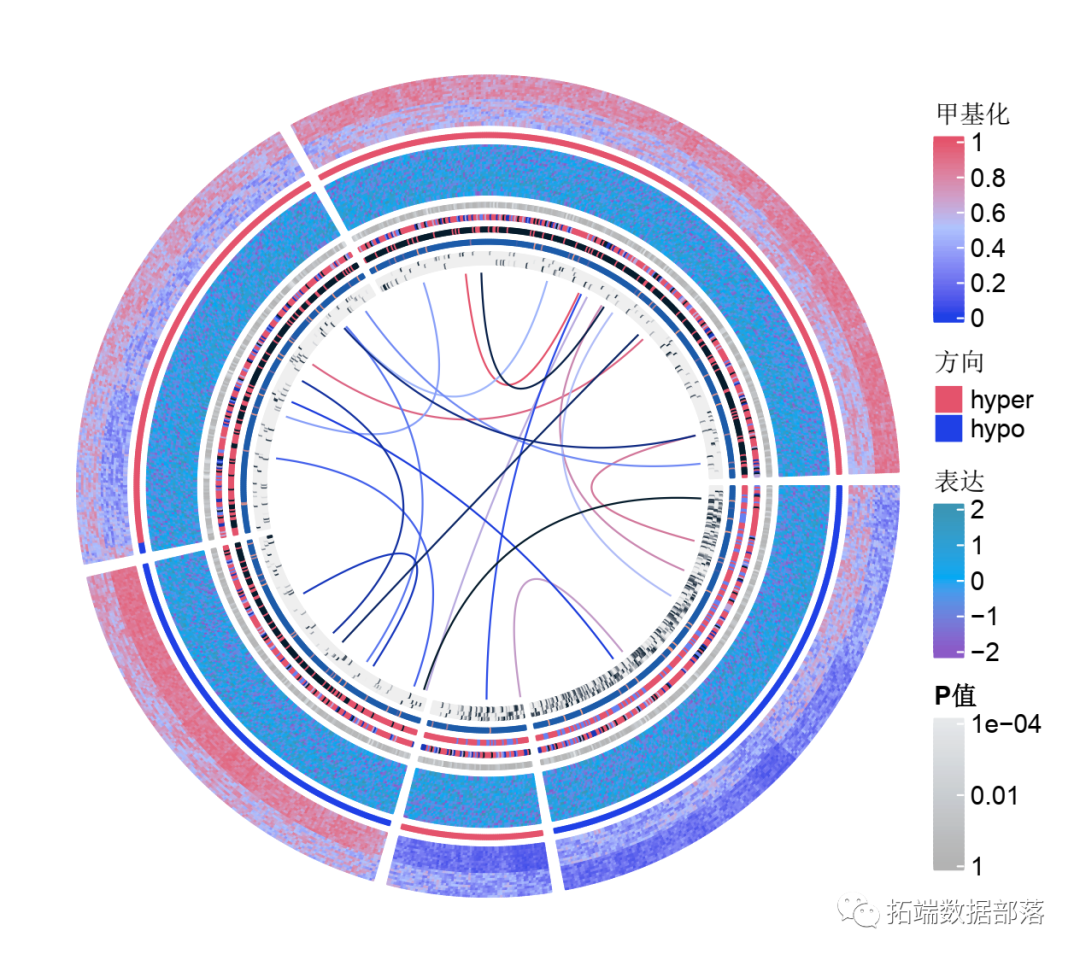
循环热图看起来很美! 由于矩阵中的行是基因组区域(差异甲基化区域),如果我们能在一些区域之间建立联系,例如三维染色体结构中的物理相互作用,那么这个图就会更漂亮、更有用。
在下面的代码中,我在DMRs之间生成一些随机的相互作用。df_link中的每一行意味着有一个从第i个DMR到第j个DMR的互动。
-
data.frame(
-
from_index
-
to_index
在圆形热图上找到这些DMR的位置是很棘手的。请看下面代码中的注释。注意这里的子集和行序元数据是通过get.data()函数明确指定扇形索引来检索的。
-
-
for(i in seq_len(nrow)) {
-
# 让我们把索引为df_link$from_index[i]的DMR称为DMR1。
-
# 另一个索引为df_link$to_index[i]的为DMR2。
-
-
# DMR1所在的扇区。
-
group1 = km[from_index[i] ]。
-
# DMR2所在的扇区。
-
group2 = km[to_index[i] ]。
-
-
# 区块`group1`中的DMRs子集(来自mat_meth的行指数)。
-
data("subset", sector.index = group1)
-
# 扇区`group1`中的行排序。
-
data("row_order", sector.index = group1)
-
# 这是DMR1在`group1`热图中的位置。
-
which(subset1[row_order1] == from_index[i])
-
-
# 区块`group2`中的DMRs子集(来自mat_meth的行指数)。
-
data("subset", sector.index = group2)
-
# 扇区`group2`中的行排序。
-
ret.data("r sector.indexoup2)
-
# 这是DMR2在`group2`热图中的位置。index[i])
-
-
# 我们取中间的点,在D和DMR2之间画一个链接
-
link(group1, x1 - 0.5, grup2,col = rcoor(1))
-
-
我实现了一个函数。现在绘制矩阵行之间的链接更简单。
-
for(i in seq_len(nrow(df_link))) {
-
heatmap.link(from[i],
-
to_index[i],
-
color(1))
-
}
添加链接后,绘图看起来更漂亮了!
图例对于理解热图非常重要。
绘制圆形图的函数只是前面代码的一个封装,没有任何修改。
图例对于理解热图非常重要。按照该链接的说明,我们需要一个绘制圆形图的函数和一个Legends对象。
现在我们使用gridBase来结合基础图形和网格图形。
-
-
-
h = dev.size()[2]
-
-
draw(lgd_list, x = circle, just = "left")

最受欢迎的见解
3.Python数据可视化-seaborn Iris鸢尾花数据
7.R语言动态可视化:制作历史全球平均温度的累积动态折线图动画gif视频图

 浙公网安备 33010602011771号
浙公网安备 33010602011771号