表面能的理解+lammps计算
1.什么是表面能?(可以去b站和youtube上看相关的介绍和解说,很多老师解释得很详细)
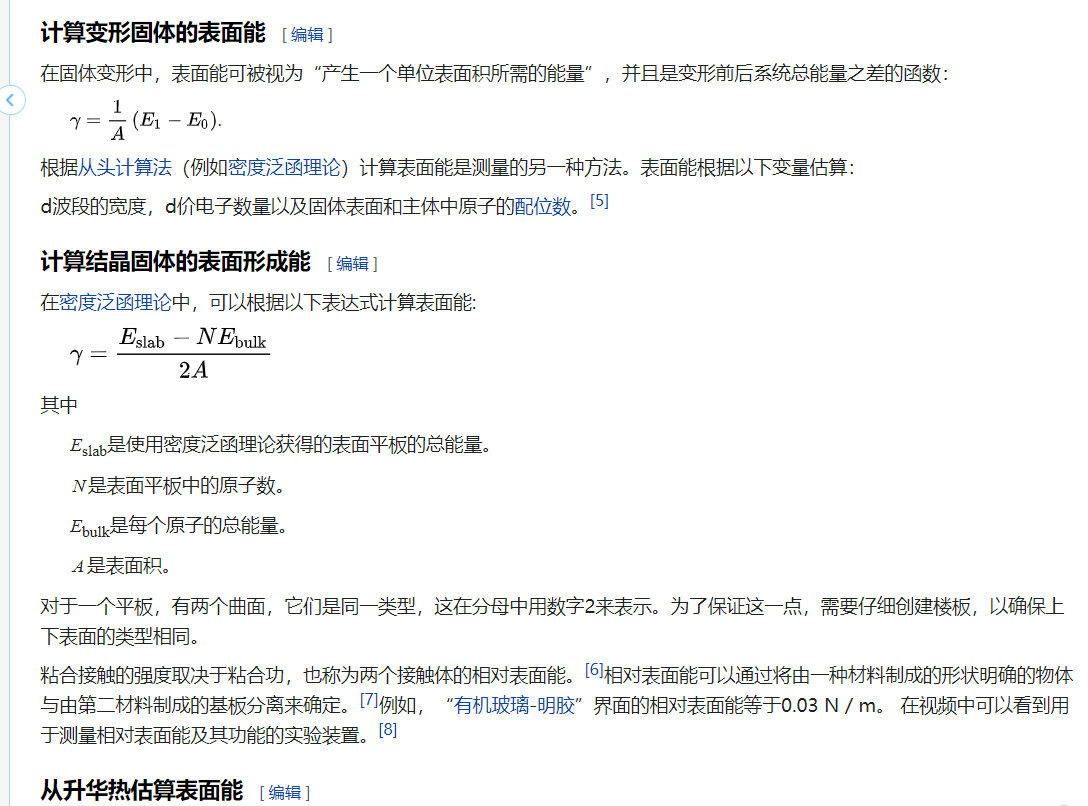
定义1:将体相变成表面晶体所需要的能量。
定义2:表面能是恒温、恒压、恒组成情况下,可逆地增加物系表面积须对物质所做的非体积功(单一相)。(对比:粘附功W12是指从相邻两相1和2之间分离需要做的功(两相),可以是液液之间或者液固相界面)
表面能的另一种定义是,表面粒子相对于内部粒子所多出的能量。表面能是创造物质表面时对分子间化学键破坏的度量。(来自360百科:https://baike.so.com/doc/2911063-3071933.html)
定义3:表面能(英语:Surface energy)是创造物质表面时,破坏分子间作用力所需消耗的能量。在固体物理理论中,表面原子比物质内部的原子具有更多的能量,因此,根据能量最低原理,原子会自发的趋于物质内部而不是表面。表面能的另一种定义是,材料表面相对于材料内部所多出的能量。把一个固体材料分解成小块需要破坏它内部的分子间作用力,所以需要消耗能量。如果这个分解的过程是可逆的,那么把材料分解成小块所需要的能量就和小块材料表面所增加的能量相等。但事实上,只有在真空中刚刚形成的表面才符合上述能量守恒。因为新形成的表面是非常不稳定的,它们通过表面原子重组和相互间的反应,或者对周围其他分子或原子的吸附,从而使表面能量降低。(转摘自:https://zh.wikipedia.org/wiki/%E8%A1%A8%E9%9D%A2%E8%83%BD)
自己的理解:当一个完整体系切开成为两边体系,进而增加两个表面,这个过程需要破坏两个新表面之间的化学键(相互作用),因此,需要额外吸收能量来完成这一过程,该能量就是表面能。
两个新体系的能量总和会大于原体系的能量,因为增加了两个表面,获得新的表面能(1.两个新表面的结构不稳定,能量比之前的要大;2.表面原子相对于内部高出的势能)。
计算方法:
1.Esurface = (U2 - U1)/2A
其中,Esurface:表面能; U2:切割成为两个体系后的总能量之和;U1:原体系的总能量;A:接触面积。
2.还有其他多种计算方式:如接触角测量法、以及如下计算方法。
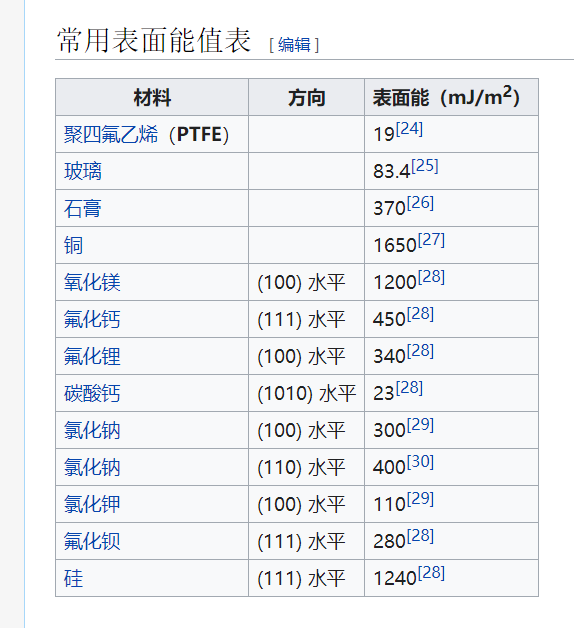

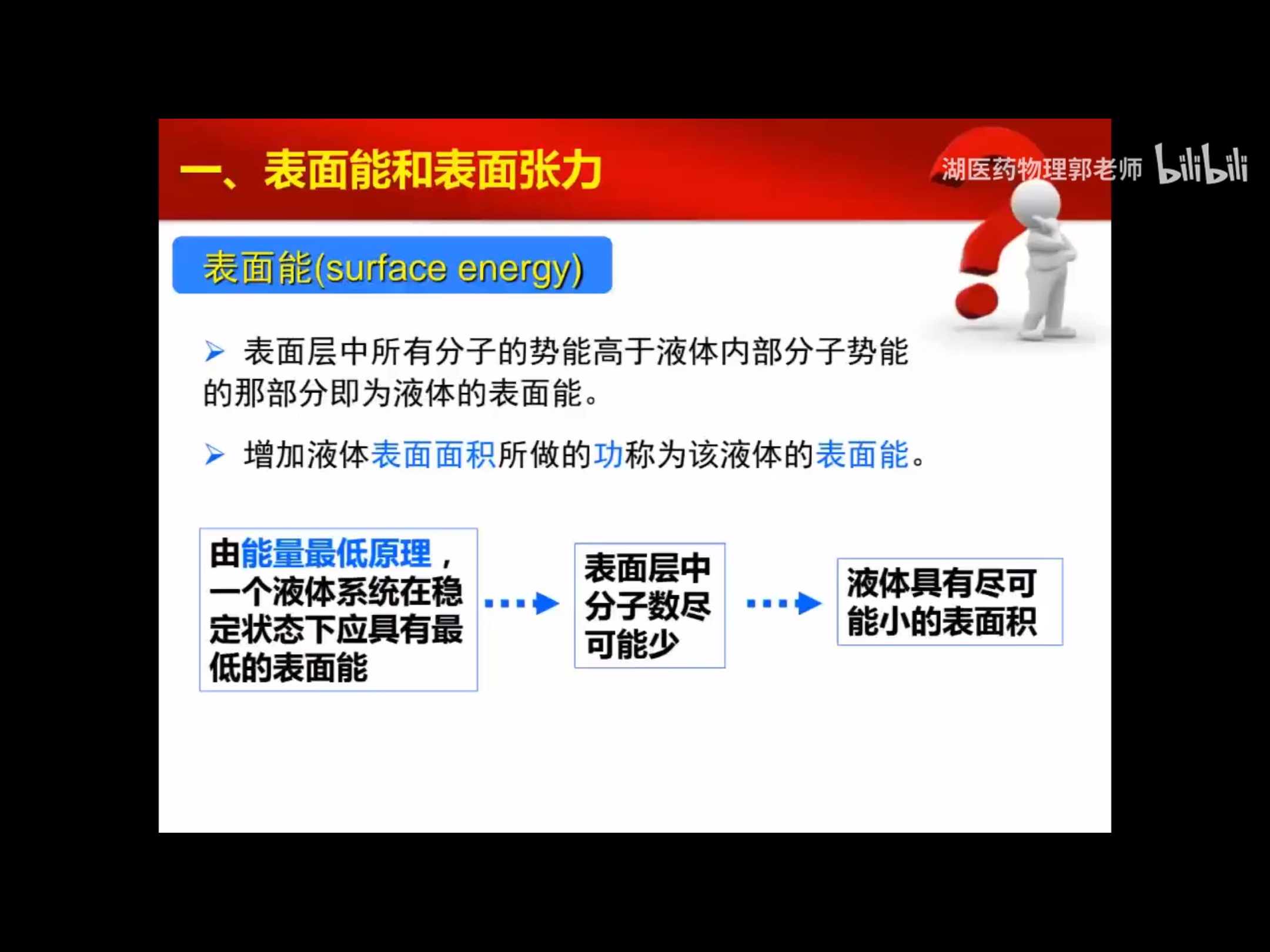
2.表面能的讨论2 (转摘自:https://www.ossila.com/en-eu/pages/a-guide-to-surface-energy)
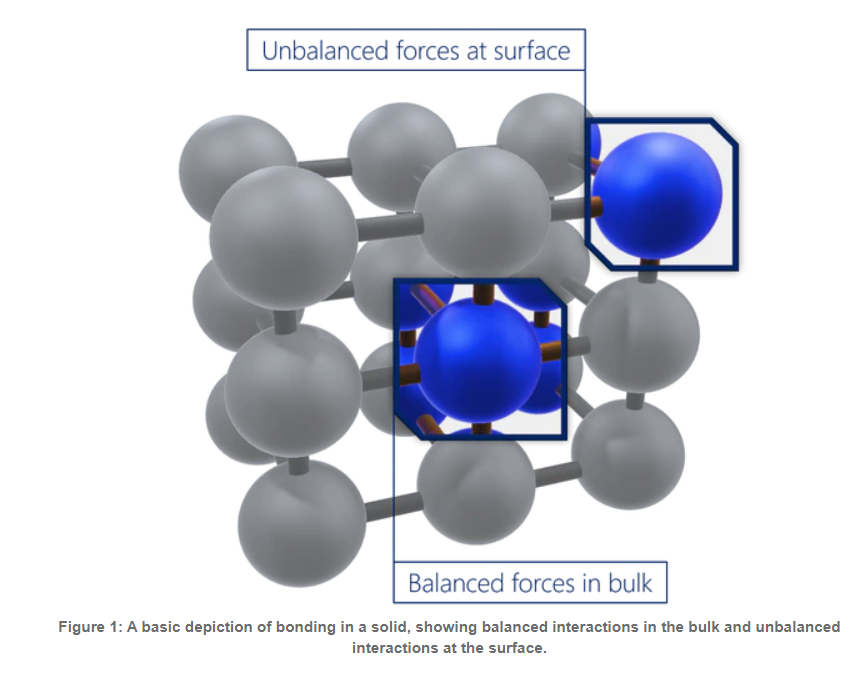
在物质的体积形态中,原子通常是稳定的,并且有一组平衡的键/相互作用。相比之下,表面原子会有一组不完整的、不平衡的相互作用,因此有未实现的键能。“表面能”是表面能量的相对测量(这是不完全结合的结果)。它与体相互作用强度和地表暴露水平呈正相关。因此,如果体相互作用更强或表面暴露程度更大,表面能就会更高。
一个表面总是会尽量减少它的能量。这可以通过将能量较低的材料吸附到其表面来实现。通过吸附过程,使表面能较高的暴露表面原子数量最小化,并被能量较低的原子或分子取代。液体的表面能通常比固体低(由于分子间的作用力较弱),这就是为什么液体通常会扩散。表面能可以定义为单位面积上增加表面尺寸所需的能量,因此经常以mN/m为单位引用。


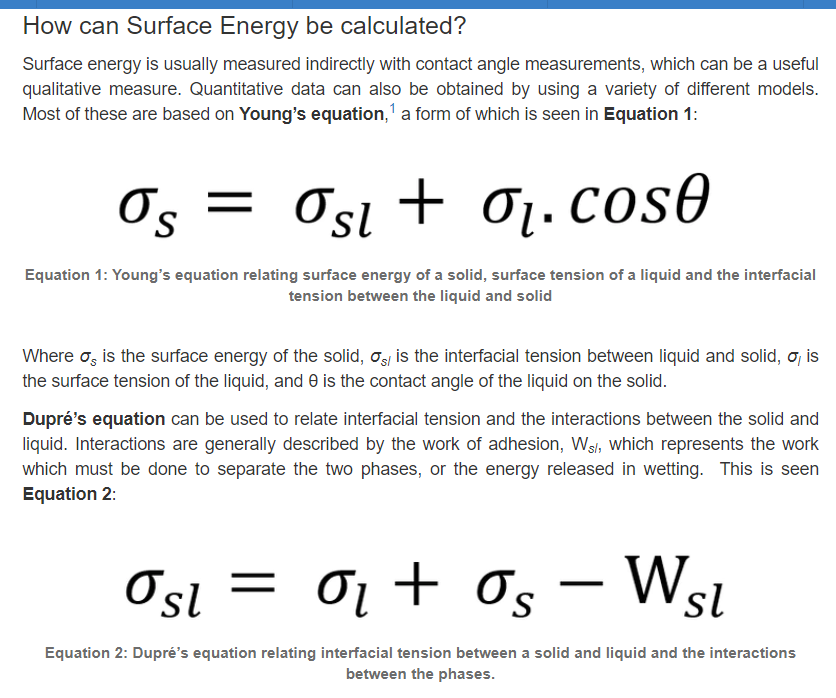
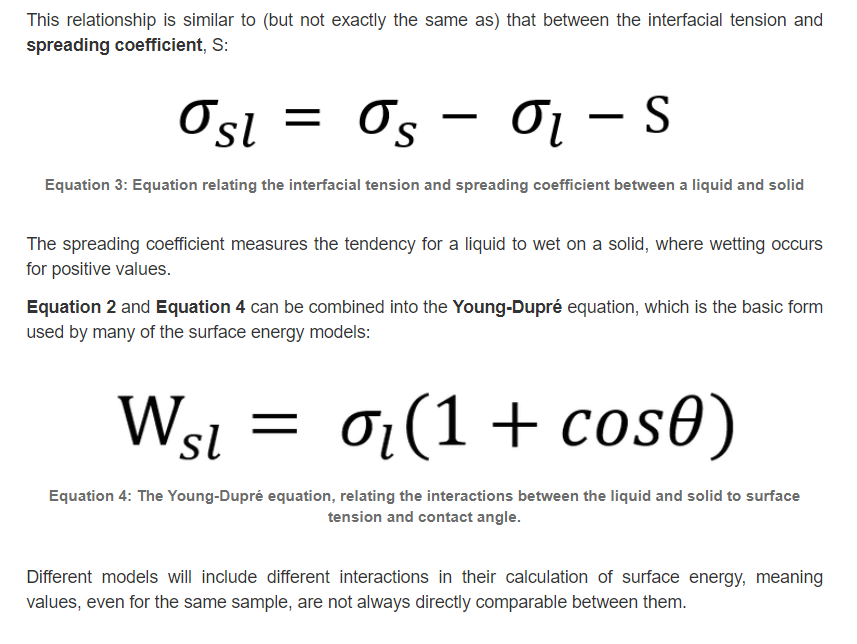
3.表面能的lammps算例,用能量最小化来计算,也有用跑平衡后计算的。根据自己的需求来计算,能量最小化是分子静力学,不考虑温度影响。
variable filename1 index P0.0001GPasurface #定义文件名
log ${filename1}.log
#模拟过程具体的in文件如下:
units real
boundary p p p #f:非周期性和固定
atom_style full
pair_style lj/cut/coul/long 10 8.5
pair_modify mix arithmetic #CLAYFF力场中的参数组合形式
pair_modify shift yes
bond_style harmonic
angle_style harmonic
kspace_style ewald 1.0e-6
read_data P0.0001GPaeq-zlength.data #MS导出的data文件
set type 1 charge 1.50133929 #ao 由1.575改为1.49507186
set type 2 charge 2.03265323 #st 由2.1改为 .... ,为了实现电荷平衡
set type 3 charge -1.05 #ob
set type 4 charge -0.95 #oh
set type 5 charge 1.0 #Na
set type 6 charge 0.425 #ho
set type 7 charge 1.36 #mgo
set type 8 charge -1.1808 #obos
set type 9 charge -1.0808 #ohs
set type 10 charge -1.1688 #obts
set type 11 charge 1.5750 #at
set type 12 charge 0.41 #h*
set type 13 charge -0.82 #o*
neighbor 1.0 bin
neigh_modify delay 0 every 1 check yes
##########################给体系划分不同的组#########################
region up block INF INF INF INF 21.6 INF #滑块最上层部分(4+顶部Na)
region down block INF INF INF INF INF 21.6 #基体最下层部分(4面体)
group up region up
group down region down
group mmt type 1 2 3 4 5 6 7 8 9 10 11
group wat type 12 13
#需要用到的组。 subtract减法、减去、删除;intersect 相交;union 加法、联合
group upmmt intersect up mmt
group downmmt intersect down mmt
##########################求出E1初体系,第一次minimize#########################
timestep 1
thermo 1000
thermo_style custom step temp vol press evdwl epair ecoul ebond eangle pe ke etotal
dump 1 all atom 1000 ${filename1}.lammpstrj
fix 1 all box/relax aniso 0.0 #盒子边长可以变化,relax,各向异性
min_style cg
minimize 1.0e-6 1.0e-8 50000 1000000
run 0
variable PE equal pe
variable E1 equal ${PE}
##########################改变体系的层状结构#########################
#change_box all z final -20 120 boundary p p p remap units box #会改变原子坐标
change_box all z delta -20 70 #往z方向扩大负-20,正70A,不改变原子坐标,但是不能读入restart文件
displace_atoms upmmt move 0 0 30 units box
#displace_atoms wat move 0 0 30 units box
write_data ${filename1}-initial.data
##########################求出E2分离后的体系,第二次minimize#########################
unfix 1
fix 2 all box/relax aniso 0.0
min_style cg
minimize 1.0e-6 1.0e-8 50000 1000000
run 0
variable A equal lx*ly
variable PE equal pe
variable E2 equal ${PE}
variable Esurf equal (${E2}-${E1})/2/$A #real单位:kcal/mol/A2
variable Esurf_ev equal ${Esurf}/23.061 #换算单位为ev/A2
print "${E2} ${E1} ${A} ${Esurf} ${Esurf_ev}" file ${filename1}.dat
write_data ${filename1}.data
write_restart ${filename1}.restart

 浙公网安备 33010602011771号
浙公网安备 33010602011771号