空间单细胞|在Seurat中对空间数据进行分析(4)
引言
在这篇指南中,我们介绍了Seurat的一个新扩展功能,用以分析新型的空间解析数据,将重点介绍由不同成像技术生成的三个公开数据集。
- Vizgen MERSCOPE(用于小鼠大脑研究)
- Nanostring CosMx空间分子成像仪(用于FFPE人类肺组织)
- Akoya CODEX(用于人类淋巴结研究)
人体淋巴结:Akoya CODEX 系统
这个数据集是通过 Akoya CODEX 系统创建的,该系统能够进行多路复用的空间分辨蛋白质分析,逐步展示抗体的结合过程。这个数据集展示了一个来自人类淋巴结的组织切片,由佛罗里达大学在人类生物分子图谱计划(HuBMAP)框架下生成。数据集中包含了28个蛋白质标记,这些蛋白质的强度是利用Akoya处理器流水线进行量化的,最终生成了一个CSV文件,该文件记录了每个细胞中各个标记的强度值以及它们的细胞位置坐标。
我们首先通过 Seurat 软件包中的 LoadAkoya() 函数来导入 HuBMAP 数据集。
codex.obj <- LoadAkoya(filename = "/brahms/hartmana/vignette_data/LN7910_20_008_11022020_reg001_compensated.csv",
type = "processor", fov = "HBM754.WKLP.262")
我们现在可以运行无监督分析来识别细胞簇。为了标准化蛋白质数据,我们使用基于中心对数比的标准化,就像我们通常应用于 CITE-seq 数据的蛋白质模态一样。然后我们运行降维和基于图的聚类。
codex.obj <- NormalizeData(object = codex.obj, normalization.method = "CLR", margin = 2)
codex.obj <- ScaleData(codex.obj)
VariableFeatures(codex.obj) <- rownames(codex.obj) # since the panel is small, treat all features as variable.
codex.obj <- RunPCA(object = codex.obj, npcs = 20, verbose = FALSE)
codex.obj <- RunUMAP(object = codex.obj, dims = 1:20, verbose = FALSE)
codex.obj <- FindNeighbors(object = codex.obj, dims = 1:20, verbose = FALSE)
codex.obj <- FindClusters(object = codex.obj, verbose = FALSE, resolution = 0.4, n.start = 1)
我们可以基于基于蛋白质强度的 UMAP 嵌入或基于它们的空间位置来可视化细胞簇。
DimPlot(codex.obj, label = TRUE, label.box = TRUE) + NoLegend()

ImageDimPlot(codex.obj, cols = "parade")

每个标记的表达模式清晰地揭示了细胞的多样性和它们在空间上的排列,例如 Lyve1 代表的淋巴内皮细胞、CD34 代表的血管内皮细胞,以及 CD21 代表的 B 细胞。正如所料,内皮细胞形成了血管结构,而 B 细胞则是构成生发中心这一特殊微环境的重要角色。在这个预印本论文中,您可以进一步了解这个数据集中的蛋白质标记详情,以及在人类淋巴组织中的细胞网络情况。
p1 <- ImageFeaturePlot(codex.obj, fov = "HBM754.WKLP.262", features = c("CD34", "CD21", "Lyve1"),
min.cutoff = "q10", max.cutoff = "q90")
p2 <- ImageDimPlot(codex.obj, fov = "HBM754.WKLP.262", cols = "parade")
p1 + p2
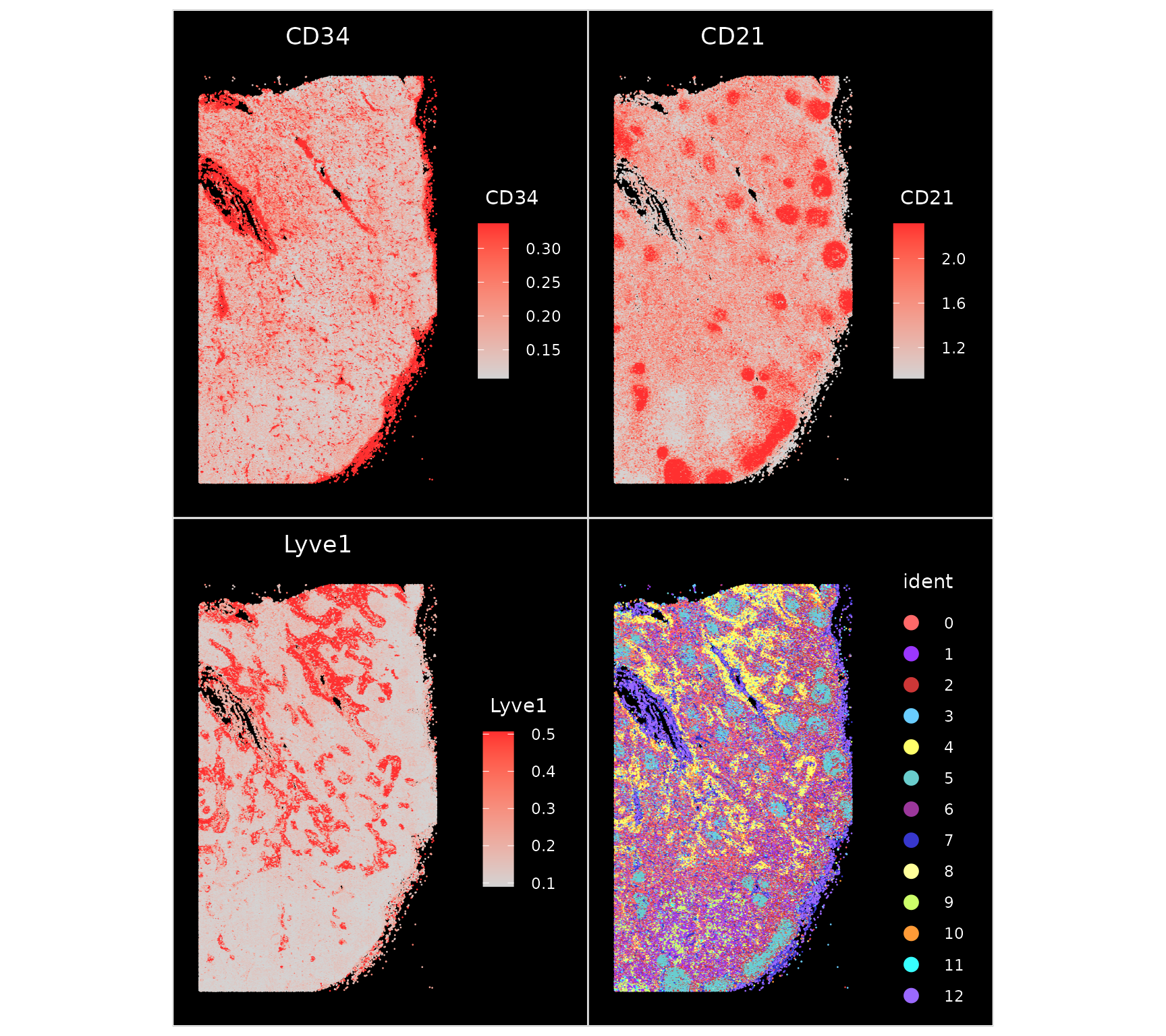
这些数据集为探索细胞在空间上如何有序分布提供了宝贵的学习机会。敬请期待 Seurat 未来版本带来的新功能,它们将帮助我们更深入地研究细胞的空间位置与其分子状态之间的联系。
本文由mdnice多平台发布

 浙公网安备 33010602011771号
浙公网安备 33010602011771号