

空间单细胞|10x Visium数据分析、可视化与整合(1)
空间单细胞|10x Visium数据分析、可视化与整合(1)
引言
本文介绍了使用Seurat分析具有空间分辨率的RNA测序数据的方法,重点在于将空间信息与分子数据相结合。将包括以下常见于空间数据分析的任务:
- 数据标准化
- 降维和数据聚类
- 发现空间变异性特征
- 与单细胞RNA测序数据的整合
- 处理多个样本切片
首先,将加载Seurat及其所需的其他包以进行本教程的操作。
library(Seurat)
library(SeuratData)
library(ggplot2)
library(patchwork)
library(dplyr)
数据集
本文将介绍一个最新发布的小鼠脑矢状面切片数据集,该数据集是利用Visium v1化学技术生成的。数据集包括两组连续的前脑切片和两组(配对的)后脑切片。
您可以利用Seurat中的Load10X_Spatial()函数将其导入。该函数能够读取spaceranger流程的输出结果,并生成一个包含点级表达数据和相应组织切片图像的Seurat对象。
此外,您还可以通过SeuratData包轻松获取数据,如下面的示例所示。安装完数据集之后,输入命令?stxBrain即可获取更多相关信息。
InstallData("stxBrain")
brain <- LoadData("stxBrain", type = "anterior1")
数据预处理
对基因表达数据的初步处理过程与一般的单细胞RNA测序实验相仿。首先,需要对数据进行标准化处理,以校正不同数据点之间测序深度的差异。发现,空间数据集在分子计数或点上的变异可能非常显著,尤其是当组织中的细胞密度不同时。在这里观察到了显著的异质性,这就需要进行有效的数据标准化。
plot1 <- VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()
plot2 <- SpatialFeaturePlot(brain, features = "nCount_Spatial") + theme(legend.position = "right")
wrap_plots(plot1, plot2)

这些图表证明了分子计数的变异不单纯是技术层面的问题,还与组织结构有关。例如,组织中神经元较少的区域(如大脑皮层的白质部分),通常会显示出较低的分子计数。因此,一些常规方法(如LogNormalize()函数),它们要求每个数据点在标准化后具有相同的“基数”,可能会引起问题。
建议改用sctransform方法(Hafemeister和Satija,2019年发表于《基因组生物学》),这种方法通过建立基因表达的正则化负二项模型,旨在消除技术误差,同时保留生物学上的变异。sctransform能够对数据进行标准化处理,识别变异性大的特征,并将这些数据保存在SCT检测项中。
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
基因表达可视化
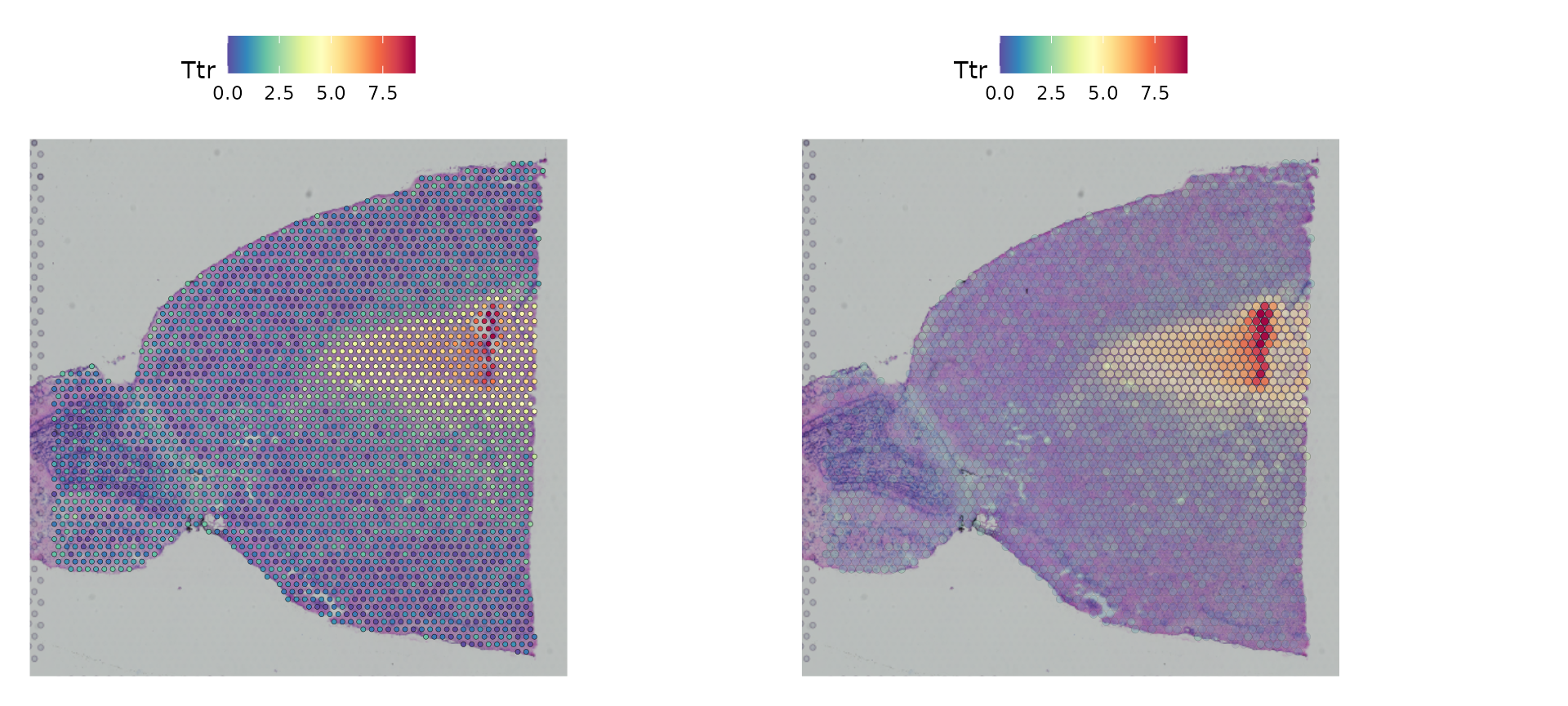
Seurat的SpatialFeaturePlot()函数是对FeaturePlot()的一个扩展,它允许在组织学图像上叠加分子层面的数据。例如,在小鼠大脑的这个数据集中,Hpca基因是海马区的一个强烈指示标志,而Ttr基因则是脉络丛的标志。
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))

library(ggplot2)
plot <- SpatialFeaturePlot(brain, features = c("Ttr")) + theme(legend.text = element_text(size = 0),
legend.title = element_text(size = 20), legend.key.size = unit(1, "cm"))
jpeg(filename = "../output/images/spatial_vignette_ttr.jpg", height = 700, width = 1200, quality = 50)
print(plot)
dev.off()
## agg_png
## 2
在Seurat中,默认设置更侧重于分子数据的可视化效果。不过,您可以通过调整一些参数来改变点的尺寸(和透明度),从而提升组织学图像的可视性:
pt.size.factor参数用于调整点的尺寸大小,其默认值为1.6。alpha参数用来设置点的透明度范围,其默认值是c(1, 1),即完全不透明到完全透明。
您可以尝试将alpha参数设置为c(0.1, 1),这样可以降低那些表达量较低的点的透明度,使得可视化效果更加突出。
p1 <- SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 1)
p2 <- SpatialFeaturePlot(brain, features = "Ttr", alpha = c(0.1, 1))
p1 + p2
本文由mdnice多平台发布
