
单细胞测序最好的教程(八): 细胞类型自动注释-1|基于marker的自动注释
作者按
本章节主要讲解了基于marker的自动注释方法,一般来说,我会先自动注释,再手动去确认marker,这是因为,对于一个陌生的组织,我对marker是不了解的,自动注释可以帮助我快速熟悉细胞类型。本教程首发于单细胞最好的中文教程,未经授权许可,禁止转载。
全文字数|预计阅读时间: 5000|5min
——Starlitnightly(星夜)
1. 背景
我们在3-2的教程中,详细介绍了如何根据marker对细胞进行手动注释的方法。但是随着时代的发展,越来越多的组织,器官被人们注释完。很多初学者对marker的不熟悉,使得其手动注释的准确率较低。这时候,我们就在想,是否可以基于现有的注释数据,构建一个全面的注释图谱,使得我们只需要输入单细胞数据,就能自动输出注释结果。
在这里,我将自动注释算法分为需要GPU的深度学习模型以及不需要GPU的统计学模型两种,我们在(一)中先介绍不需要GPU的统计学模型自动注释。我们在此介绍的算法是SCSA,SCSA可以根据簇特异性marker,查询数据库中每一种细胞类型的marker,选出吻合度最高的一类细胞,如果差异性不够大,那么则选出两类细胞,这时可以被认为是Unknown细胞。
import omicverse as ov
print(f'omicverse version: {ov.__version__}')
import scanpy as sc
print(f'scanpy version: {sc.__version__}')
ov.ov_plot_set()
2. 加载数据
在这里,我们使用手动注释好的所用到的人类骨髓数据进行注释,使用注释好的数据可以方便我们比较手动注释与自动注释的准确率。
adata = ov.read('s4d8_manual_annotation.h5ad')
adata
3. 聚类
与手动注释类似,我们使用SCSA模型也需要对单细胞测序数据先进行聚类,在这里,我们的resolution=2可以设置地更大一些。
sc.pp.neighbors(adata, n_neighbors=15,
n_pcs=30,use_rep='scaled|original|X_pca')
sc.tl.leiden(adata, key_added="leiden_res1", resolution=2.0)
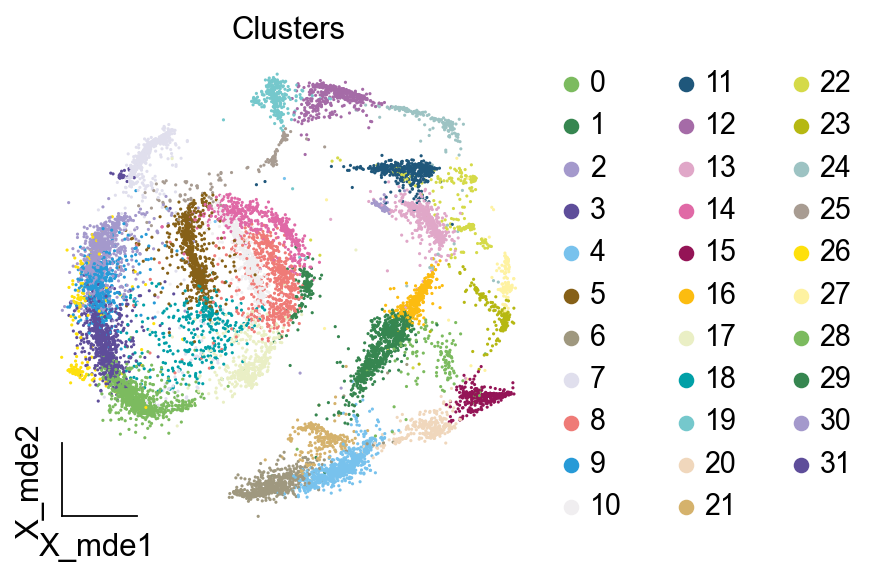
我们观察不同类别在UMAP图上的分布情况
adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
--
ov.utils.embedding(adata,
basis='X_mde',
color=[ "leiden_res1"],
title=['Clusters'],
palette=ov.palette()[:],
show=False,frameon='small',)
4. SCSA自动注释
4.1 Cellmarker注释
很多paper里,将SCSA作为一个benchmark,认为其注释准确率较低,但实际这跟SCSA所依赖的数据库有关,作者一直将CellMarker数据库停留在1.0版本,直到今年omicverse将SCSA整合,并更新数据库后,这个19年发表的算法也更新了CellMarker数据库。而在omicverse中,我还添加了panglaodb作为第二个参考数据库供读者选择。
我们在这里详细介绍函数ov.single.pySCSA的参数:
- foldchange: 这是我们每个簇相对于别的簇的差异倍数,一般设置1.5即可,设置地越高用到的marker越少
- pvalue: 与foldchange对应,我们在计算差异倍数的时候会进行统计学差异显著性分析,通常由于细胞数的原因,pvalue会出现膨胀,所以p值都非常小,我们设置一个常见阈值0.01即可
- celltype: 这个参数包括了
normal和cancer两个参数,当我们设置成cancer的时候,我们可以注释出来自cancersea数据库的12种肿瘤细胞亚型 - target: 我们要使用的目的数据库,目前pySCSA中存放了
cellmarker,cancersea,panglaodb三个数据库 - tissue: 我们可以使用
scsa.get_model_tissue()列出所有支持的组织,默认是使用全部tissue - model_path: 这里可以设定我们使用的数据库,默认为'',将自动从figshare下载omicverse专用数据库,当然你也可以手动下载
注意事项:
celltype='cancer',target='cancersea'需要同时设定
数据库:
pySCSA_2023_v2_plus.db
scsa=ov.single.pySCSA(adata=adata,
foldchange=1.5,
pvalue=0.01,
celltype='normal',
target='cellmarker',
tissue='All',
model_path='temp/pySCSA_2023_v2_plus.db'
)
正式开始注释的环节,我们这里有三个参数
- clustertype: 注释依据的簇名,存放在adata.obs
- cluster: 需要注释的cluster,可以设置为list: ['1','2']表示只注释簇1和簇2
- rank_rep: 是否需要重新计算差异表达基因,由于当细胞数量上去后,差异表达基因的计算会较慢,如果我们已经运算了
sc.tl.rank_genes_groups,我们可以设置成False停止重复计算
adata.uns['log1p']['base']=10
res=scsa.cell_anno(clustertype='leiden_res1',
cluster='all',rank_rep=True)
res.head()
我们可以使用cell_anno_print来打印自动注释的结果
scsa.cell_anno_print()
Cluster:0 Cell_type:Natural killer cell|T cell Z-score:13.273|9.396
Nice:Cluster:1 Cell_type:B cell Z-score:17.404
Cluster:2 Cell_type:Natural killer cell|T cell Z-score:11.593|6.514
Cluster:3 Cell_type:Natural killer cell|T cell Z-score:12.76|8.216
Nice:Cluster:4 Cell_type:B cell Z-score:12.598
Cluster:5 Cell_type:Natural killer cell|T cell Z-score:9.54|8.235
Nice:Cluster:6 Cell_type:B cell Z-score:17.663
Nice:Cluster:7 Cell_type:Natural killer cell Z-score:14.884
Cluster:8 Cell_type:T cell|CD4+ T cell Z-score:11.135|5.887
Nice:Cluster:9 Cell_type:Natural killer cell Z-score:11.839
Cluster:10 Cell_type:T cell|Natural killer cell Z-score:8.536|5.758
Nice:Cluster:11 Cell_type:Monocyte Z-score:14.702
Nice:Cluster:12 Cell_type:Natural killer T (NKT) cell Z-score:16.376
Cluster:13 Cell_type:Monocyte|Natural killer T (NKT) cell Z-score:14.982|12.323
Cluster:14 Cell_type:T cell|Naive CD4+ T cell Z-score:4.286|3.828
Nice:Cluster:15 Cell_type:B cell Z-score:15.455
Cluster:16 Cell_type:Natural killer T (NKT) cell|B cell Z-score:16.348|14.152
Cluster:17 Cell_type:Natural killer T (NKT) cell|T cell Z-score:12.837|11.628
Cluster:18 Cell_type:T cell|Natural killer cell Z-score:12.177|9.216
Nice:Cluster:19 Cell_type:Red blood cell (erythrocyte) Z-score:12.92
Nice:Cluster:20 Cell_type:B cell Z-score:15.318
Nice:Cluster:21 Cell_type:B cell Z-score:13.077
Cluster:22 Cell_type:Monocyte|Natural killer T (NKT) cell Z-score:12.582|6.915
Nice:Cluster:23 Cell_type:B cell Z-score:16.847
Cluster:24 Cell_type:Hematopoietic stem cell|Natural killer T (NKT) cell Z-score:10.647|10.385
Nice:Cluster:25 Cell_type:Red blood cell (erythrocyte) Z-score:2.196
Nice:Cluster:26 Cell_type:Natural killer cell Z-score:14.092
Nice:Cluster:27 Cell_type:B cell Z-score:14.312
Nice:Cluster:28 Cell_type:B cell Z-score:15.943
Cluster:29 Cell_type:T cell|Naive CD8+ T cell Z-score:7.808|6.713
Cluster:30 Cell_type:Monocyte|Macrophage Z-score:12.19|8.303
Nice:Cluster:31 Cell_type:Natural killer cell Z-score:11.614
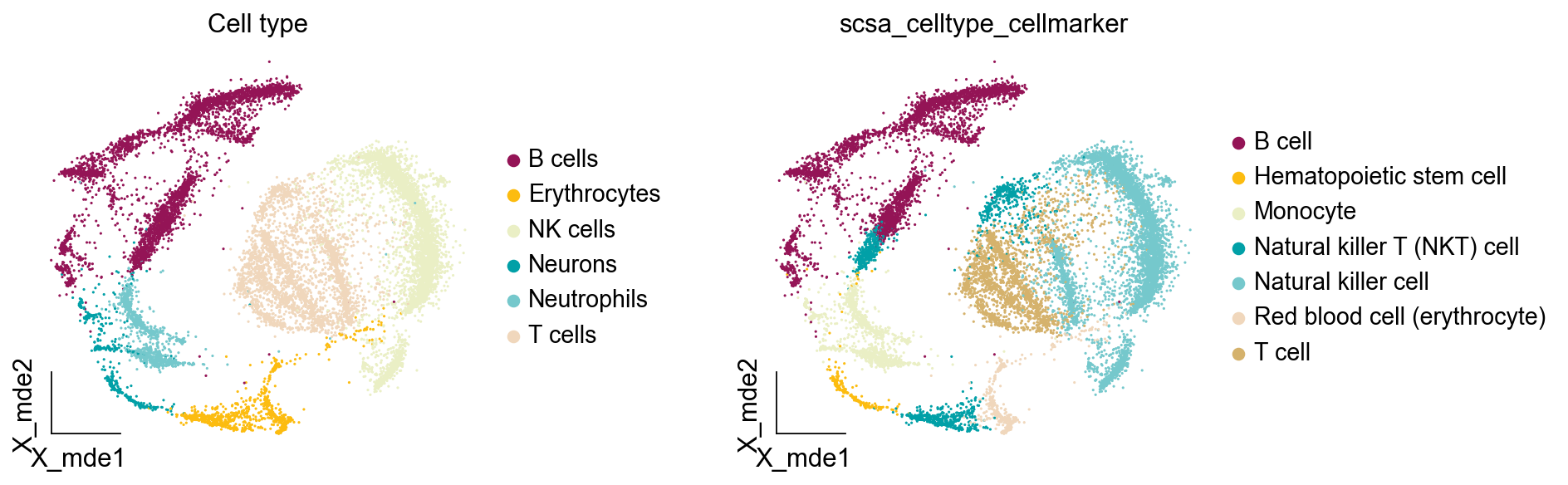
我们使用cell_auto_anno将分数最高的细胞类型注释上,但这个函数不会区分非Nice的细胞注释结果,还存在一些误差。
scsa.cell_auto_anno(adata,clustertype='leiden_res1',
key='scsa_celltype_cellmarker')
...cell type added to scsa_celltype_cellmarker on obs of anndata
我们发现自动注释中多注释出了Hematopoietic stem cell,这类细胞可能是注释偏差,其对应的簇为24,我们发现其z-score没有高于第二类两倍,10.647|10.385。此外,我们自动注释没有注释出neuron,neuron对应的是胶质细胞,这是自动注释的局限性
ov.utils.embedding(adata,
basis='X_mde',
color=[ "major_celltype","scsa_celltype_cellmarker",],
title=['Cell type'],
palette=ov.palette()[15:],
show=False,frameon='small',wspace=0.45)
4.2 panglaodb注释
我们还可以尝试panglaodb数据库来注释
scsa=ov.single.pySCSA(adata=adata,
foldchange=1.5,
pvalue=0.01,
celltype='normal',
target='panglaodb',
tissue='All',
model_path='temp/pySCSA_2023_v2_plus.db'
)
res=scsa.cell_anno(clustertype='leiden_res1',
cluster='all',rank_rep=True)
res.head()
scsa.cell_auto_anno(adata,clustertype='leiden_res1',
key='scsa_celltype_panglaodb')
...cell type added to scsa_celltype_panglaodb on obs of anndata
ov.utils.embedding(adata,
basis='X_mde',
color=[ "scsa_celltype_panglaodb","scsa_celltype_cellmarker",],
title=['Cell type'],
palette=ov.palette()[15:],
show=False,frameon='small',wspace=0.45)
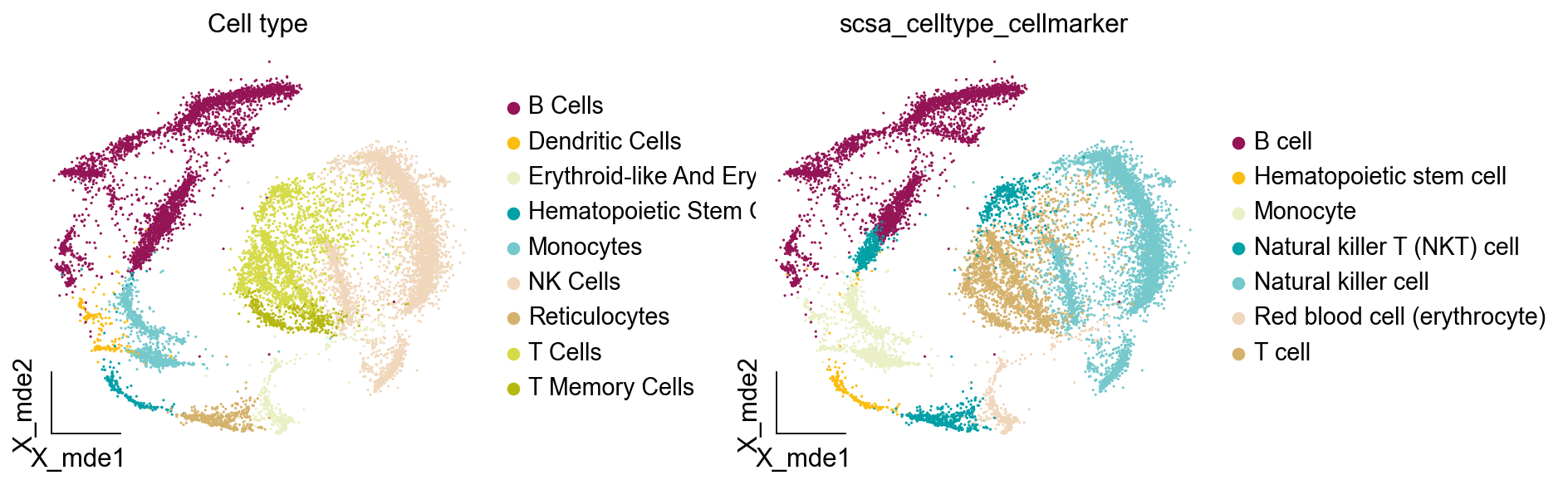
有趣的是,panglaodb注释出了Reticulocytes,Erythroid-like两种红细胞,T cells,T memory cells两种T细胞亚群。虽然看起来更准确了,但实际上我们在大类注释时不需要将细胞类型精细化。我们还提供了一个函数get_celltype_marker获取指定细胞类型的marker基因
marker_dict=ov.single.get_celltype_marker(adata,clustertype='scsa_celltype_cellmarker')
marker_dict.keys()
--
dict_keys(['B cell', 'Hematopoietic stem cell', 'Monocyte', 'Natural killer T (NKT) cell', 'Natural killer cell', 'Red blood cell (erythrocyte)', 'T cell'])
--
marker_dict['B cell']
array(['AFF3', 'BACH2', 'CD74', 'EBF1', 'BANK1', 'RALGPS2', 'IGHM', 'LYN',
'FCHSD2', 'ZCCHC7', 'PAX5', 'MEF2C', 'UBE2E2'], dtype=object)
我们可以查询所有支持的tissue,目前支持两种物种:Human与Mouse。我们在后续的更新中会陆续加入斑马鱼,植物等数据库
scsa.get_model_tissue(species='Human')
本文来自博客园,作者:Starlitnightly,转载请注明原文链接:https://www.cnblogs.com/starlitnightly/p/18258594

