4.数据质控、清洗、去接头
使用sratoolkit的fastq-dump命令转换后得到的fastq文件可以接着使用fastqc和multiqc进行质控,fastq可以对数据出具HTML报告,multiqc可以将不同数据样本的HTML报告进行整合成一个总的HTML文件,便于查看测序数据的质量;
1.对数据进行质控分析
1 #!/bin/bash 2 3 #使用fastqc对数据进行清洗 4 mkdir fastqc 5 for i in ./fastq/SRR* 6 do 7 fastqc -o "./fastqc" $i 8 9 done 10 11 #使用multiqc整合报告 12 source activate rna_py3_7 13 multiqc -o "./fastqc" ./fastqc 14 15 #将所有的报告文件打包便于下载到本地查看 16 tar -zcvf ./fastqc/qc.tar.gz ./fastqc/*.html
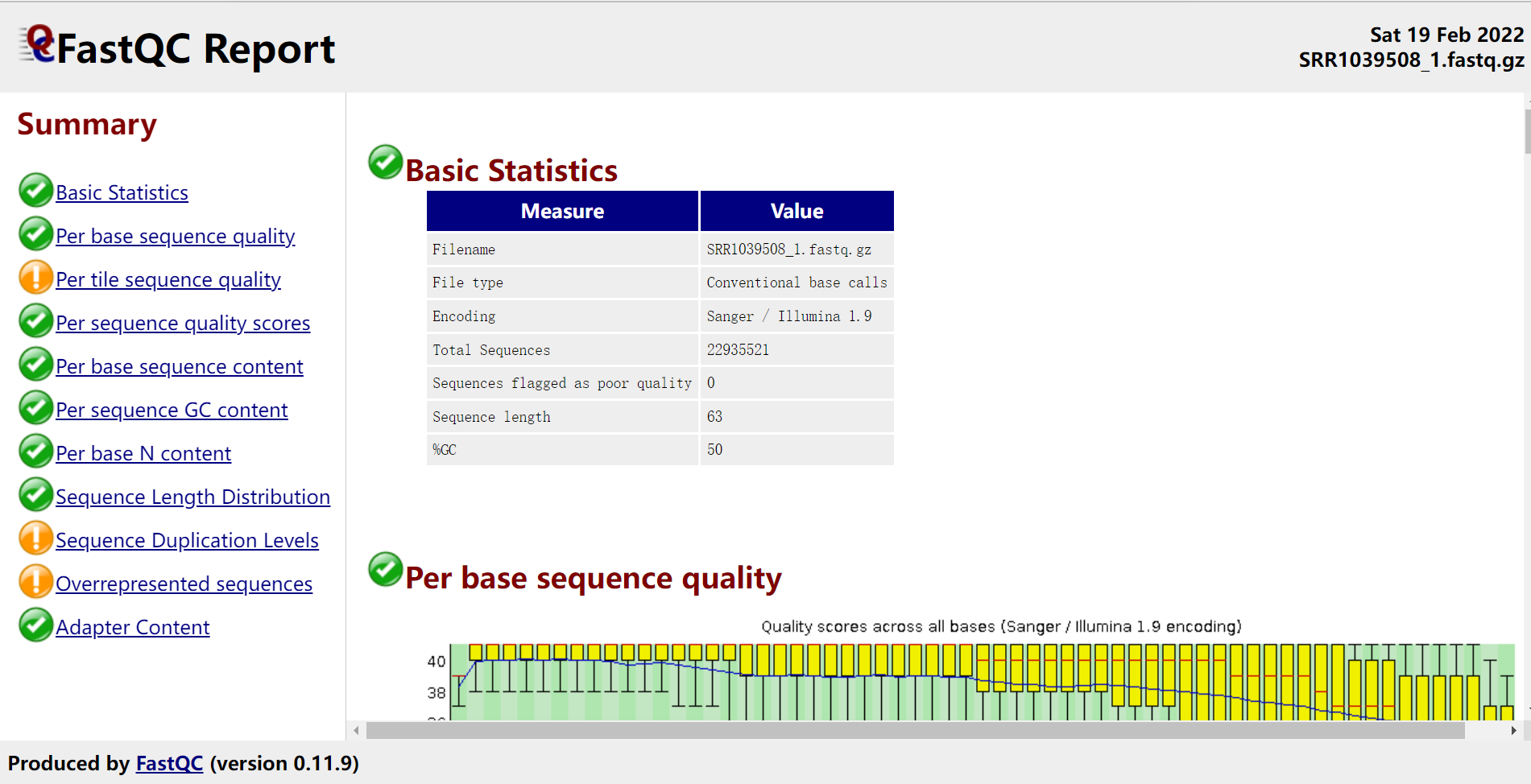
fastq出具的报告如下:
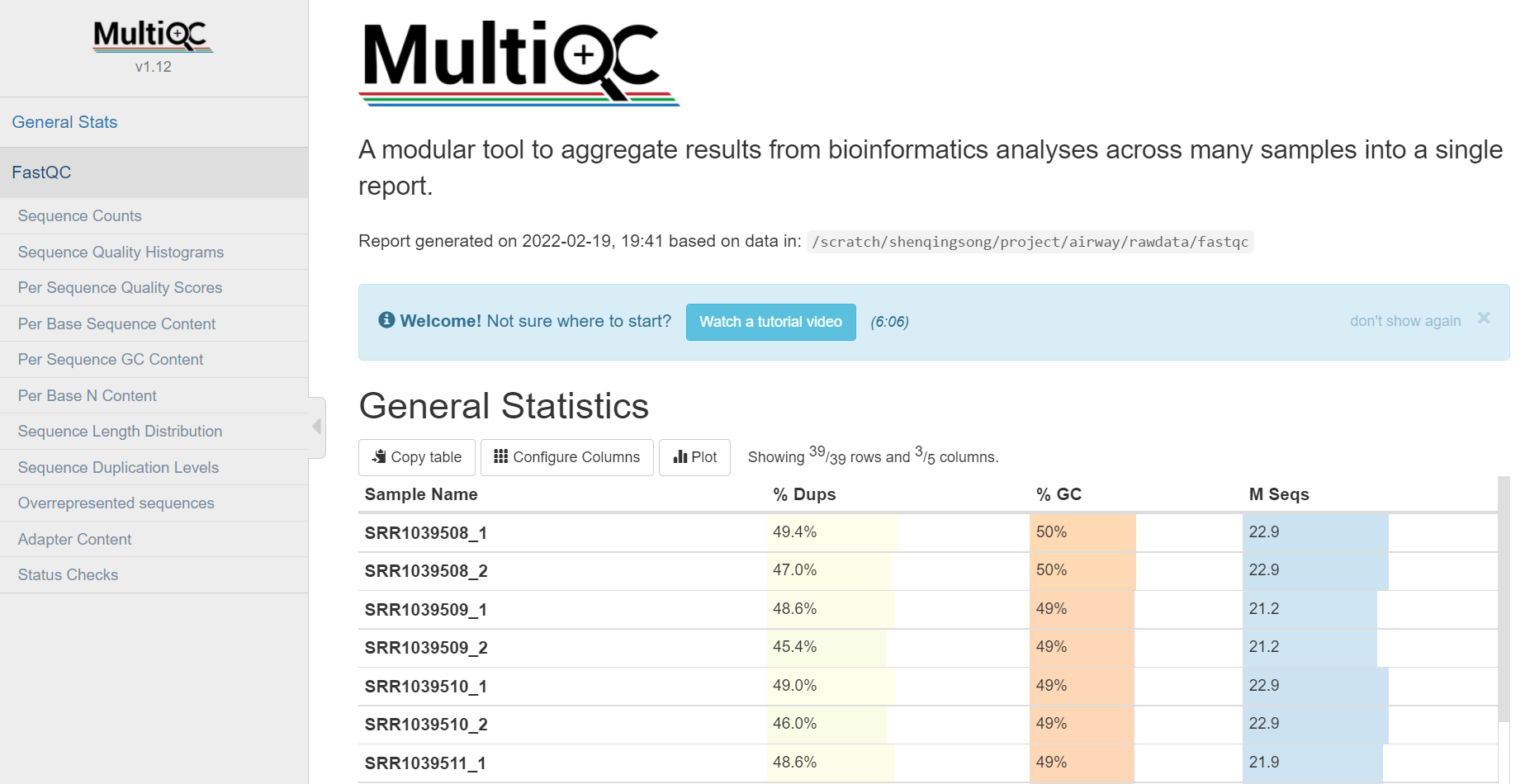
multiqc整合后的报告如下:
2.使用trim_galore对数据进行清洗去接头
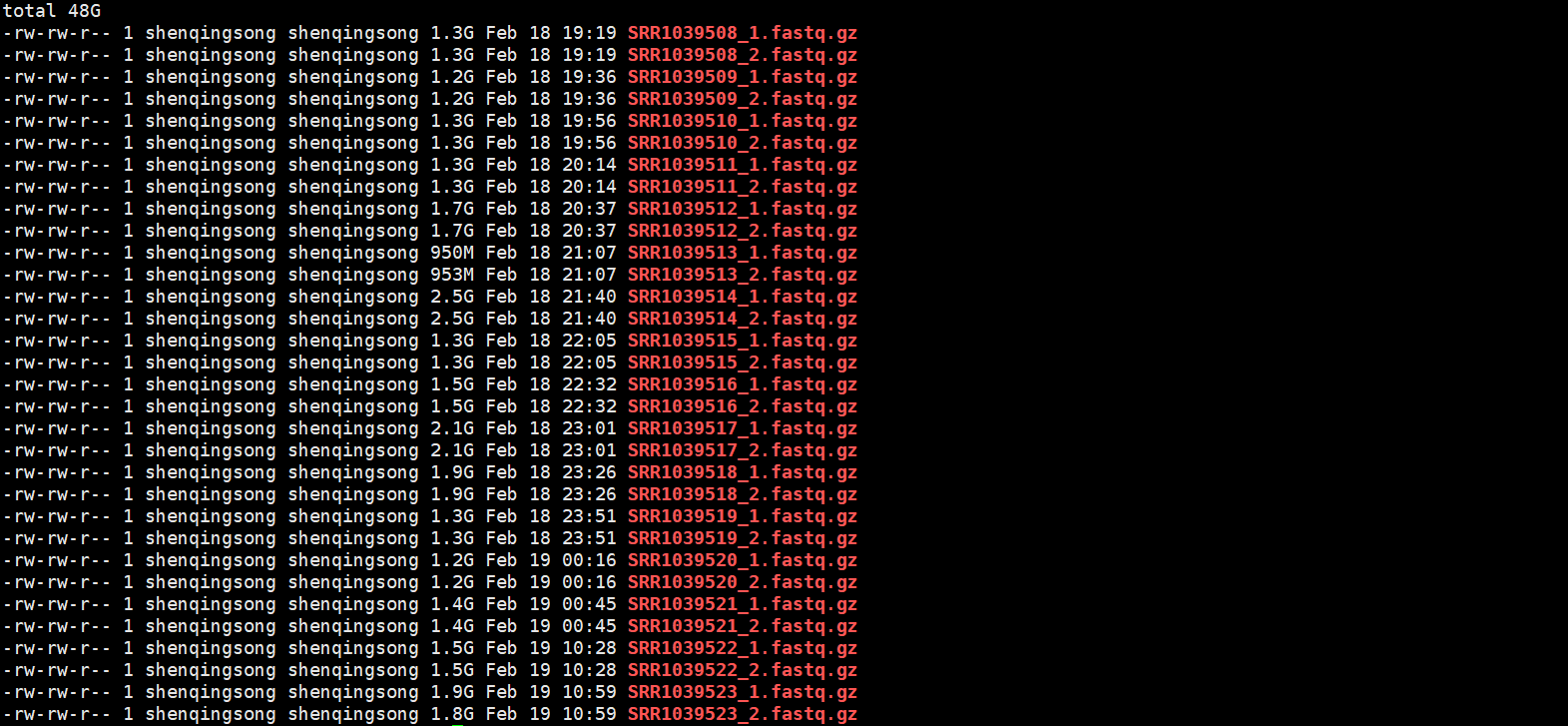
1 #查看数据储存文件夹结构 2 ll -h ./fastq
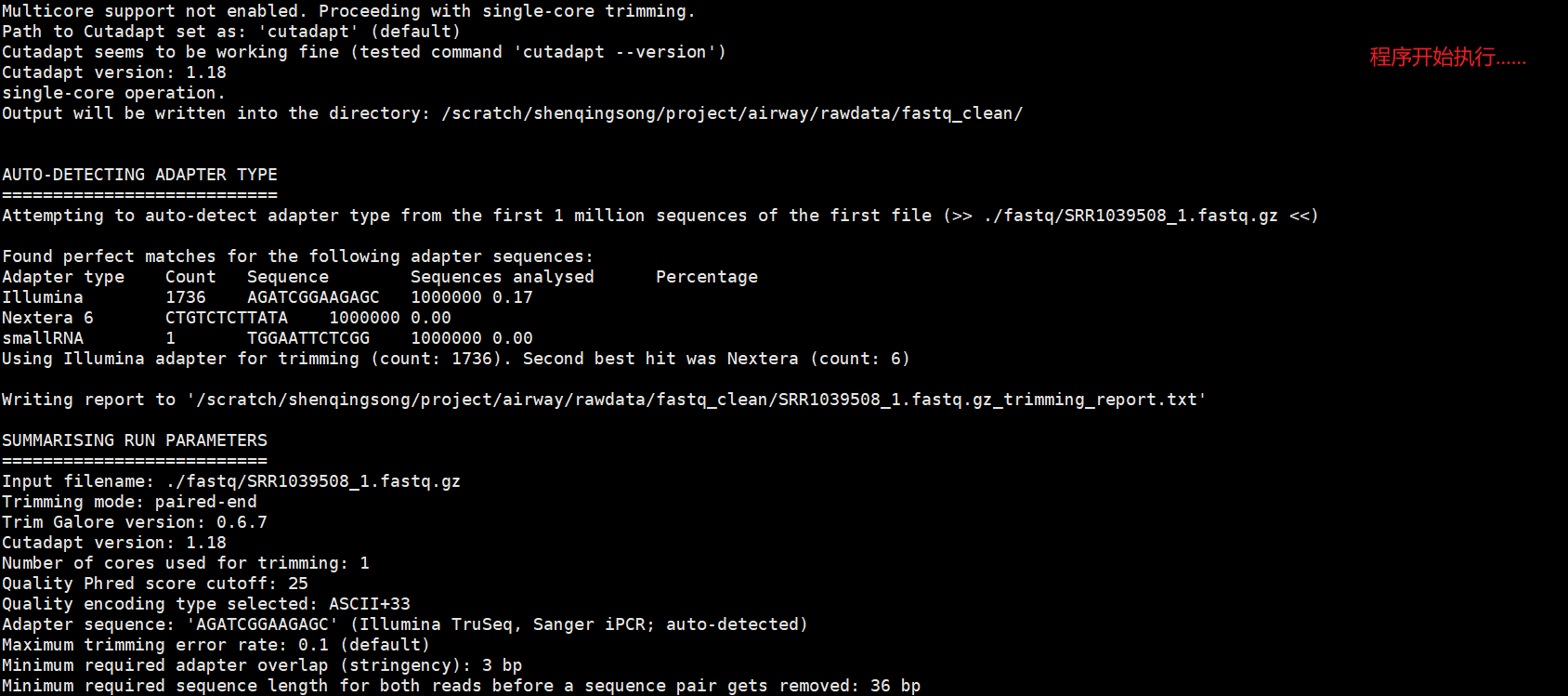
trim_galore脚本:
1 #!/bin/bash 2 3 #预处理文件名 4 ls fastq | grep "_1" > gz1 5 ls fastq | grep "_2" > gz2 6 paste gz1 gz2>config_file 7 8 #循环处理文件 9 cat config_file | while read id 10 do 11 sample_dir="./fastq" 12 output_dir="./fastq_clean" 13 arr=($id) 14 fq1=${arr[0]} 15 fq2=${arr[1]} 16 sample_dir1="$sample_dir/$fq1" 17 sample_dir2="$sample_dir/$fq2" 18 #为防止结束终端时命令挂掉,可以用nohub防止挂起,因为现在使用学校超算中心,所以不用担心该问题 19 trim_galore -q 25 --phred33 --length 36 -e 0.1 --stringency 3 --paired -o $output_dir $sample_dir1 $sample_dir2 20 done

 浙公网安备 33010602011771号
浙公网安备 33010602011771号