


AI医疗高精尖!基于AI的新药研发!⛵
 『人工智能+新药研发』已经成为国内外医药企业的发展新模式!本文讲解 AI 在新药研发领域的诸多应用方向与 MolSearch 工具库的应用实践——药物晶型预测、靶点选择、患者招募、虚拟药物筛选、AI新药研发辅助系统。
『人工智能+新药研发』已经成为国内外医药企业的发展新模式!本文讲解 AI 在新药研发领域的诸多应用方向与 MolSearch 工具库的应用实践——药物晶型预测、靶点选择、患者招募、虚拟药物筛选、AI新药研发辅助系统。

💡 作者:韩信子@ShowMeAI
📘 机器学习实战系列:https://www.showmeai.tech/tutorials/41
📘 深度学习实战系列:https://www.showmeai.tech/tutorials/42
📘 本文地址:https://www.showmeai.tech/article-detail/329
📢 声明:版权所有,转载请联系平台与作者并注明出处
📢 收藏ShowMeAI查看更多精彩内容

💡 引言&背景
💦 AI + 新药研发
人工智能作为一种新兴技术,是新药研发实现降本增效的重要方式之一,『人工智能+新药研发』成为国内外医药企业加速创新转型的重要驱动力,一个更快、更便宜、更有效的新药物研发时代已经到来。
一款新药从确认研发目标到完成临床试验,往往需要耗费数年时间和数十亿美元,并伴随着超过 90% 的失败概率。高昂的研发成本、漫长的研发周期、高风险低回报率的特性,笼罩在药物研发领域。而正在探索的各种 AI 应用,可以帮助解决这些挑战。
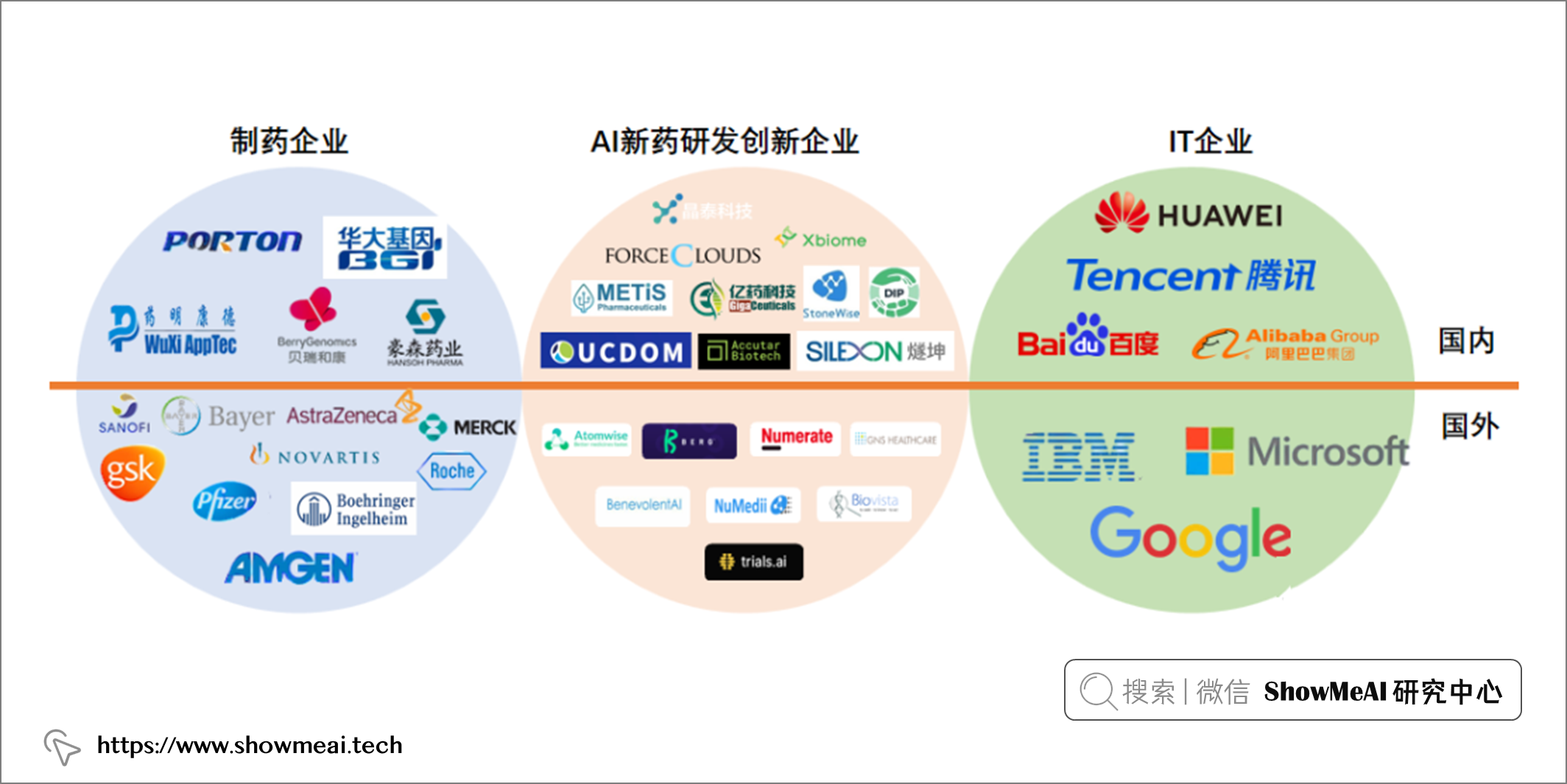
📘最近发表的一项分析表明,150 多种小分子药物处于研发阶段,超过 15 种药物已经在临床试验中,这条 AI 生物技术赛道以每年近 40% 的速度急速扩张种。为追赶这波浪潮,制药公司正在建立自己的内部人工智能团队,或与 IT 公司、AI新药研发创新公司进行投资和合作。
💦 AI 药物分子结构分析/检索
利用 AI 进行药物化合物分子结构分析和检索等,是一个助力新药研发的可行思路。Zilliz 公司与全球顶尖制药研发企业共同开发的『MolSearch』化合物分子结构分析软件是一个典型的例子。本篇咱们就一起来了解下这个过程是如何完成的。
💡 向量搜索 & 医疗研发领域的应用
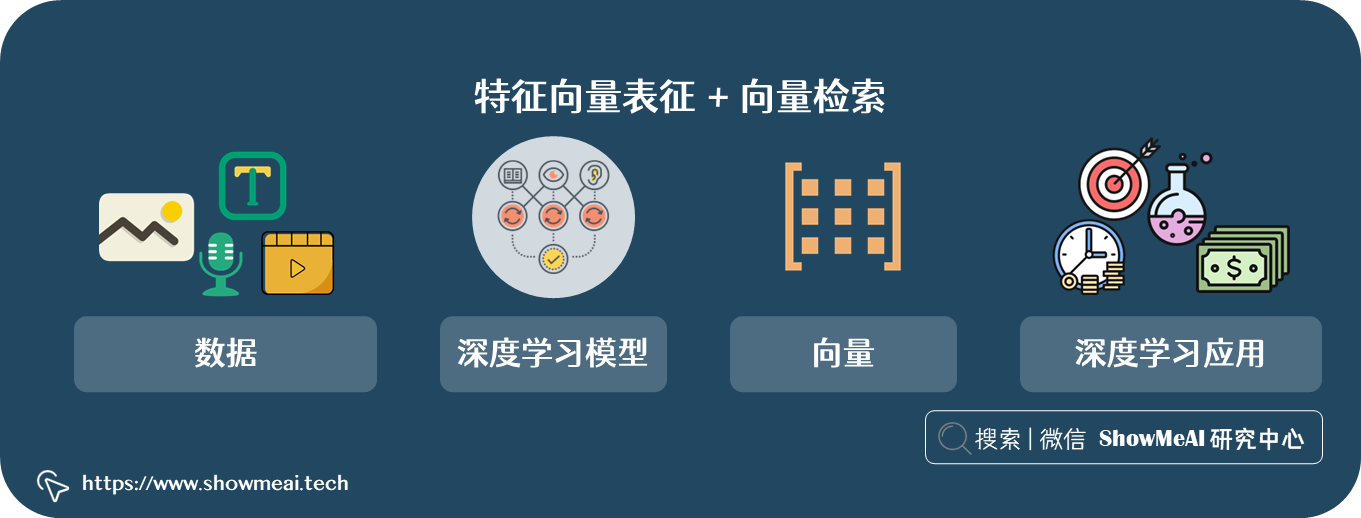
在万物皆可 embedding 的深度学习时代,『特征向量表征+向量检索』有巨大作用,在很多数据和业务领域都发挥了巨大作用,例如机器视觉(图片视频检索)、自然语言处理(文本检索、问答)、语音识别等。同样的思路也可以用在医疗医药领域。
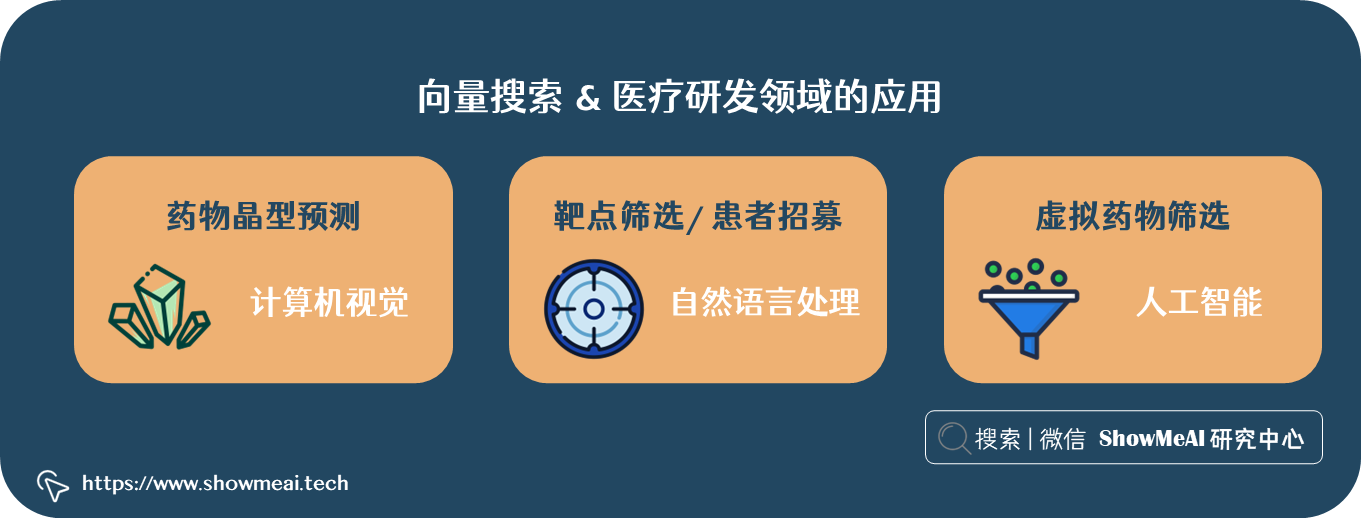
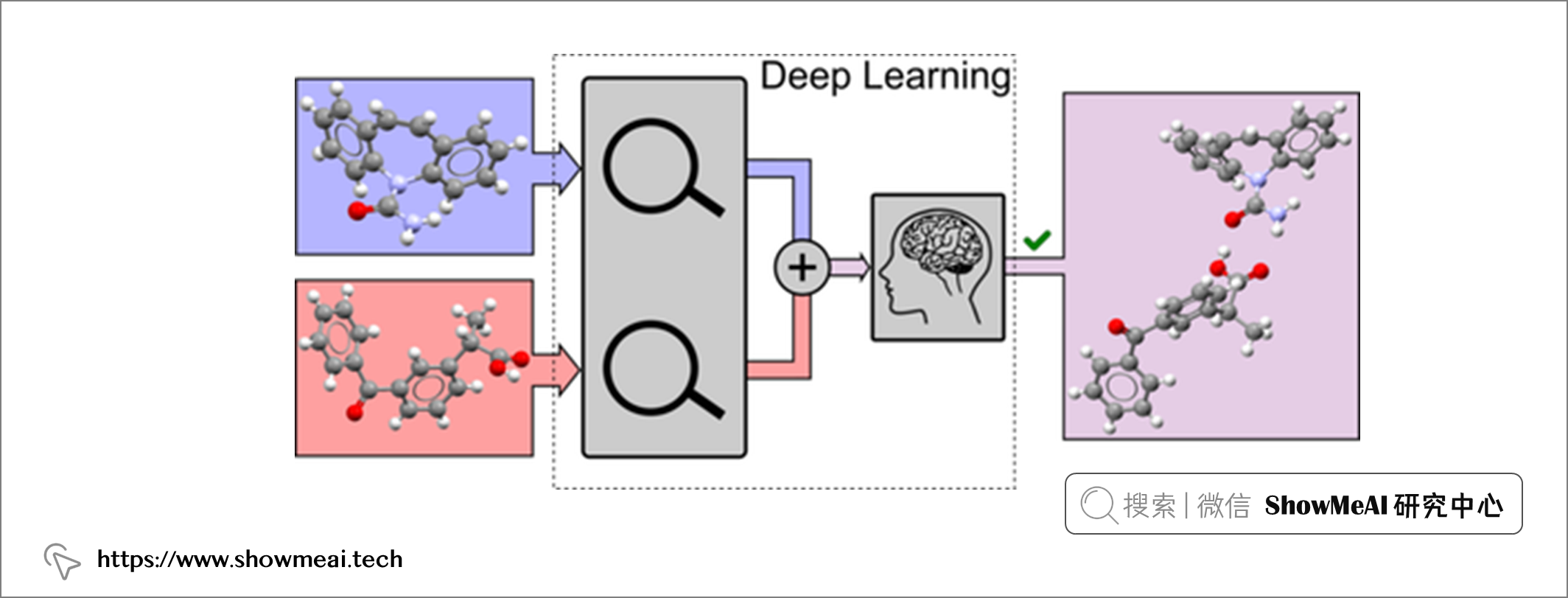
💦 药物晶型预测
比如新药研发过程药物晶型预测,可以结合图像识别和检索的思路,有效地预测出合适的药物晶型。
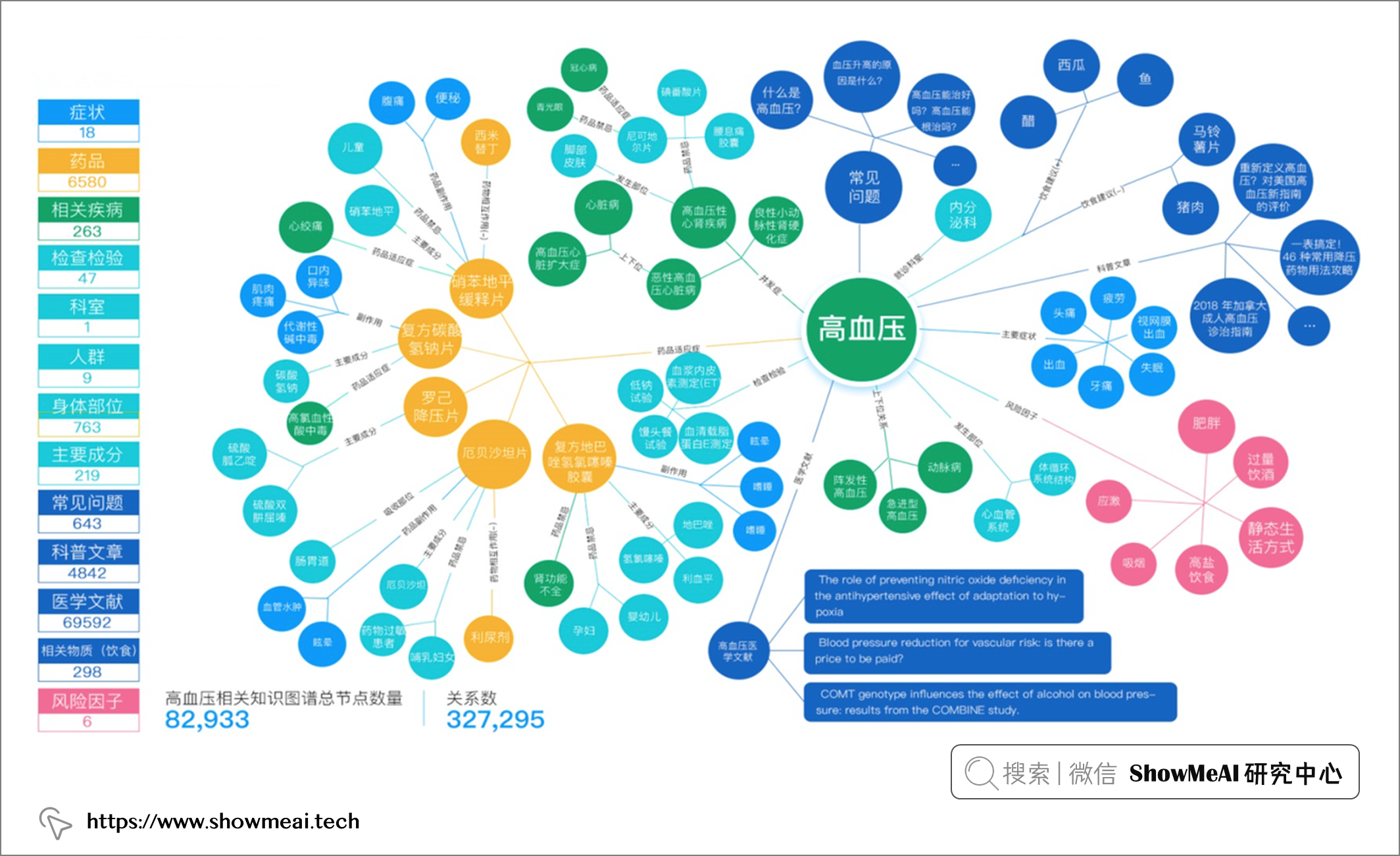
💦 靶点筛选与患者招募
比如靶点筛选和患者招募过程,可以抽象为对文本语义分析问题,可以结合 NLP 表征与检索方法,快速分析有关药物研发的文本数据等。
💡 虚拟药物筛选
AI 可以在新药研发过程中发挥巨大作用的另一个步骤是『虚拟药物筛选』,通过模拟药物筛选的过程,预测化合物可能的活性,对比较有可能成为药物的化合物进行针对性的实体筛选,这个过程可以大大降低药物研发的时间和经济成本。
有不少传统方法方案在尝试,但受限于算法和算力,对千万级别的化合物分子进行相似性、子结构、超结构等分析时,耗时较长(分钟级别),而在AI向量检索技术优化后,能大大加速这个过程(对十亿级的化学式数据极速分析,仅秒级别)。
💦 MolSearch 新药结构筛选
Zilliz 公司基于 Milvus 向量相似度检索引擎,研发了化合物分析软件 📘MolSearch,大家可以在 📘这里 查看中文说明。

药物化学专家通常根据骨架跃迁对分子模块进行优化,并基于它设计出新药结构并做后续筛选。针对海量化合物的虚拟筛选是非常核心关键的一部,其效果很大程度决定了后期小白鼠实验以及临床试验能否成功,候选底库量级越大,筛选准确率越高,新药研发成功的概率也相应越高。
MolSearch 系统集成向量相似度检索引擎 Milvus ,构建分子检索功能,可以实现十亿级的化学分子结构秒级检索分析能力。
💦 MolSearch 效果&性能
目前 MolSearch 中集成了 8.2 亿 zinc 开放化学式分子式数据集,这些化学式被转换为 2048 位的化学指纹(特征向量),在有表征特征向量之后,借助于高效向量检索引擎,可以实现对分子结构的相似性、子结构和超结构检索。

MolSearch 端到端的检索性能数据如图所示(图中『响应时间(p99)』表示 99% 的检索能在多少时间完成)。
💡 AI 新药研发辅助系统
💦 筛选流程 & 核心步骤
详细展开 MolSearch 的虚拟化合物筛选技术如下图所示,包含以下步骤:
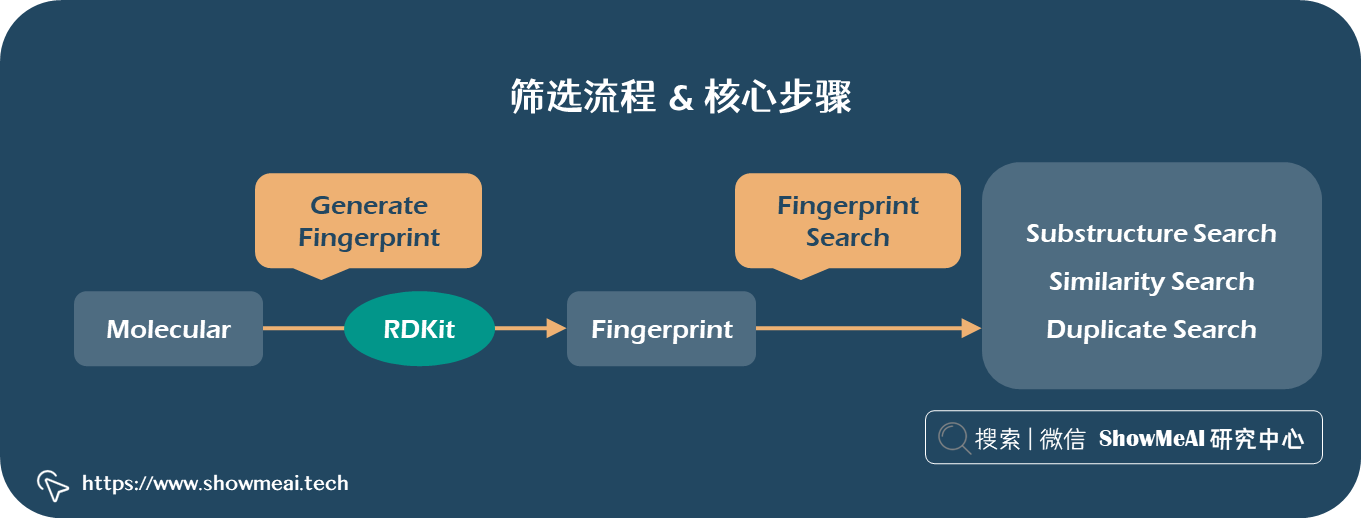
- ① 通过 📘RDKit 工具将化合物分子的化学式转换为化学式指纹/Chemical Fingerprint(也即表征特征向量)。
- ② 通过向量检索引擎,对化合物分子之间关系分析:子结构检索、相似性检索、重复结构检索。
💦 化学指纹生成
化学指纹通常用来做结构检索和相似度检索,如下图所示,最终的指纹向量表征为01串,每一位(0/1)代表化学结构中例如指定元素,分子片段等是否存在。
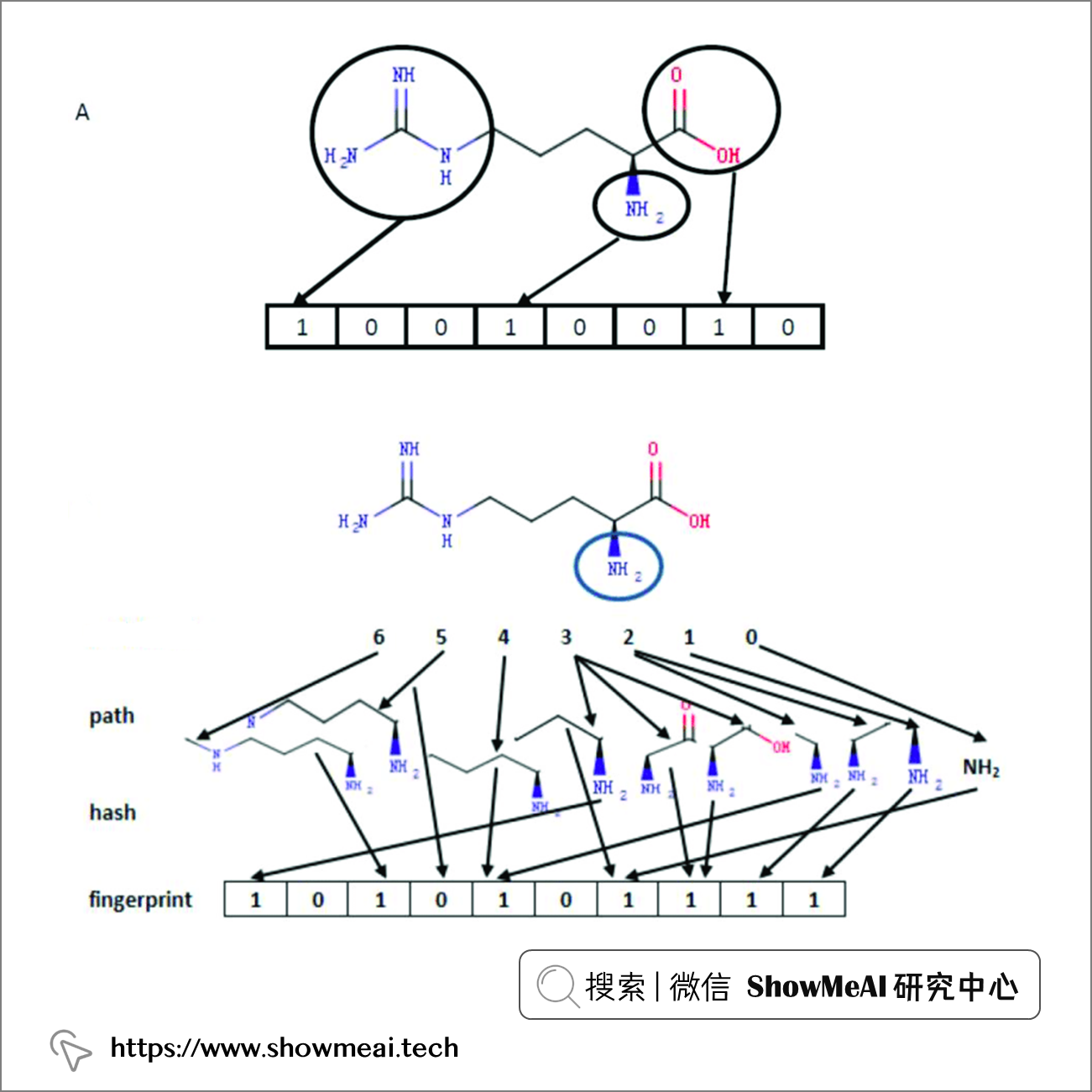
MolSearch 中这个环节使用了工具 RDKit ,它会生成 RDKit fingerprint,底层的算法原始是:分析从一个原子开始直至到达指定数量键的路径(path,通常为线性)上所有的分子片段,然后对每一个路径进行哈希(hash)产生指纹(fingerprint)。
上图展示了从NH2(已圈出)开始一直到 6 个长度的所有路径,然后将每个路径 hash 映射为二进制位。
图例是一个单个起始原子出发的片段和比特位,最终的完整指纹生成,是对分子中的每个原子进行这个操作后的结果。可以指定 fpSize 调整生成的向量维度,这个过程对于每个分子都适用,我们把最终生成的向量导入 Milvus 以实现后续检索,完整的指纹向量生成过程示例代码如下:
from rdkit import Chem
mols=Chem.MolFromSmiles(smiles)
fp=Chem.RDKFingerprint(mols,fpSize=VECTOR_DIMENSION)
bit_fp=DataStructs.BitVectToFPSText(fp)
vectors=bytes.fromhex(hex_fp)
💦 化合物检索
我们将生成的指纹向量导入 Milvus,即可应用不同计算方式完成对化合物的『相似度检索』、『子结构检索』和『超结构检索』。示例代码如下:
from milvus import *
milvus = Milvus()
milvus.insert(collection_name=MILVUS_TABLE, records=vectors)
milvus.search(collection_name=MILVUS_TABLE, query_records=query_list, top_k=topk, params={})
- 相似度检索。用于寻找与输入的参考分子比较相似的分子。
- 子结构检索。检测一个分子结构是否为另一个分子的子结构。
- 超结构检索。检测一个分子结构是否为另一个分子的超结构。
💦 指纹距离度量与相似度计算
Milvus工具本身支持各种常用相似度计算指标,包括『欧氏距离』、『内积』、『汉明距离』和『Jaccard距离』等。因为指纹是二值型数据向量,我们可以选择 Jaccard/Substructure(子结构)/Superstructure(超结构) 距离计算相似度。我们定义以下表示:

根据以上定义,化学式指纹之间的距离和相似度度量计算,可以如下表中描述来计算:

参考资料
- 📘 AI in small-molecule drug discovery: a coming wave?:(https://www.nature.com/articles/d41573-022-00025-1
- 📘 MolSearch 官方 GitHub:https://github.com/zilliztech/MolSearch
- 📘 MolSearch 中文说明:https://github.com/zilliztech/MolSearch/blob/master/CN_README.md
- 📘 RDKit:https://www.rdkit.org/

