为什么 Biopython 的在线 BLAST 这么慢?
用过网页版本 BLAST 的童鞋都会发现,提交的序列比对往往在几分钟,甚至几十秒就可以得到比对的结果;而通过调用 API 却要花费几十分钟或者更长的时间!这到底是为什么呢?

NCBIWWW 基本用法
首先,我们来看一下提供了基于 API 在线比对的 Biopython 模块。
Biopython 中的 BLAST 提供了 over the Internet 和 locally 两种选择:Bio.Blast.NCBIWWW 主要是基于 NCBI BLAST API 用于在线比对;Bio.Blast.Applications 模块则是调用本地安装好的 BLAST 程序以及数据库执行比对。在这里我们来重点看一下 Bio.Blast.NCBIWWW 。
Bio.Blast.NCBIWWW 模块中主要是通过 qblast() 函数来调用 BLAST 的在线版本。它具有三个非可选参数:
第一个参数是用于搜索的 blast 程序,为小写字符串。目前,
qblast(biopython==1.7.4)仅适用于 blastn,blastp,blastx,tblast 和 tblastx。第二个参数指定要搜索的数据库。关于这个选项,在 NCBI Guide to BLAST 上有详细的描述。
第三个参数是包含查询序列的字符串。这可以是序列本身,也可以是 fasta 格式的序列,或者是诸如 GI 号之类的标识符。
qblast 函数还接受许多其他选项参数,这些参数基本上类似于我们可以在 BLAST 网页上设置的不同参数。我们在这里只重点介绍其中一些:
参数
url_base是设置用于在 Internet 上运行 BLAST 的基本 URL。默认情况下,它连接到 NCBI(即 url_base='https://blast.ncbi.nlm.nih.gov/Blast.cgi'),但是可以使用它连接到云端运行的 NCBI BLAST 实例。更多详细信息请参阅 qblast 功能的文档。qblast 函数可以返回各种格式的 BLAST 结果,您可以使用可选的
format_type关键字进行选择:“HTML”,“Text”,"ASN.1” 或 "XML"。默认值为 “XML”,因为这是解析器期望的格式。参数
expect用于设置期望值或 e-value 阈值。
>>> from Bio.Blast import NCBIWWW
>>> help(NCBIWWW.qblast)
请注意,NCBI BLAST 网站上的默认设置与 qblast 上的默认设置不太相同。如果获得不同的结果,则需要检查参数(例如,e-value 值和 gap 值)。
>>> from Bio.Blast import NCBIWWW
>>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
>>> from Bio.Blast import NCBIWWW
>>> fasta_string = open("m_cold.fasta").read()
>>> result_handle = NCBIWWW.qblast("blastn", "nt", fasta_string)
SeqRecord 对象进行读取,然后仅提供序列本身进行比对:
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.seq)
SeqRecord 对象的
format 方法来制作 FASTA 字符串(其中将包含现有标识符):
>>> from Bio.Blast import NCBIWWW
>>> from Bio import SeqIO
>>> record = SeqIO.read("m_cold.fasta", format="fasta")
>>> result_handle = NCBIWWW.qblast("blastn", "nt", record.format("fasta"))
无论给 qblast() 函数提供什么参数,都应在 handle 对象(默认为 XML 格式)中返回结果。下一步是将 XML 输出解析为表示搜索结果的 Python 对象,但是您可能想先保存输出文件的本地副本。在调试从 BLAST 结果中提取信息的代码时,我发现这特别有用(因为重新运行在线搜索速度很慢,并且浪费了 NCBI 计算机时间)。
result_handle.read() 读取一次 BLAST 输出——再次调用
result_handle.read() 会返回一个空字符串。
>>> with open("my_blast.xml", "w") as out_handle:
... out_handle.write(result_handle.read())
...
>>> result_handle.close()
>>> result_handle = open("my_blast.xml")
现在我们已经将 BLAST 结果重新放回了句柄中,下一步,如果我们准备对它们进行处理,我们可以参考 Biopython 中 Parsing BLAST output 部分的内容,这里不再说明。
NCBIWWW 实现
在了解 NCBIWWW 的实现前,我们先来看一下 NCBI BLAST 对于 API 使用的一些说明:
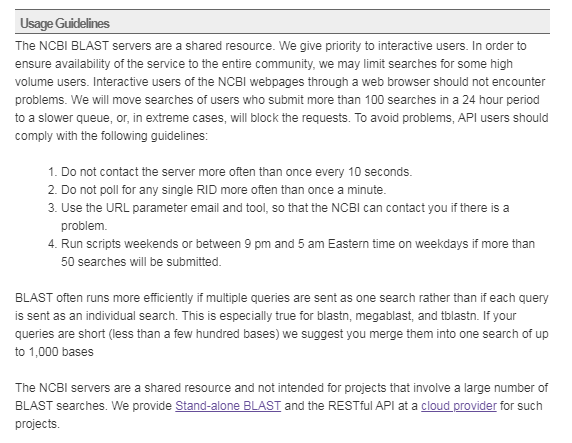
NCBI BLAST 服务器是共享资源。为了确保整个社区都能使用该服务,他们可能会限制某些高流量用户的搜索。
他们会将在 24 小时内提交 100 次以上搜索的用户的搜索移到较慢的队列中,或者在极端情况下将阻止请求。
NCBI BLAST 优先考虑互动的用户,通过网络浏览器的 NCBI 网页的交互式用户不会遇到以上的问题。
对于 API 的使用准则:
与服务器联系的频率不要超过每 10 秒一次。
不要轮询每一个 RID(Request ID) 多于一分钟一次。
使用 URL 参数电子邮件和工具,以便 NCBI 在出现问题时可以与您联系。
如果将提交超过 50 个搜索,则在周末或东部时间东部时间晚上 9 点至凌晨 5 点之间运行脚本。
我们再来看一下 NCBIWWW 在源码层面的处理:
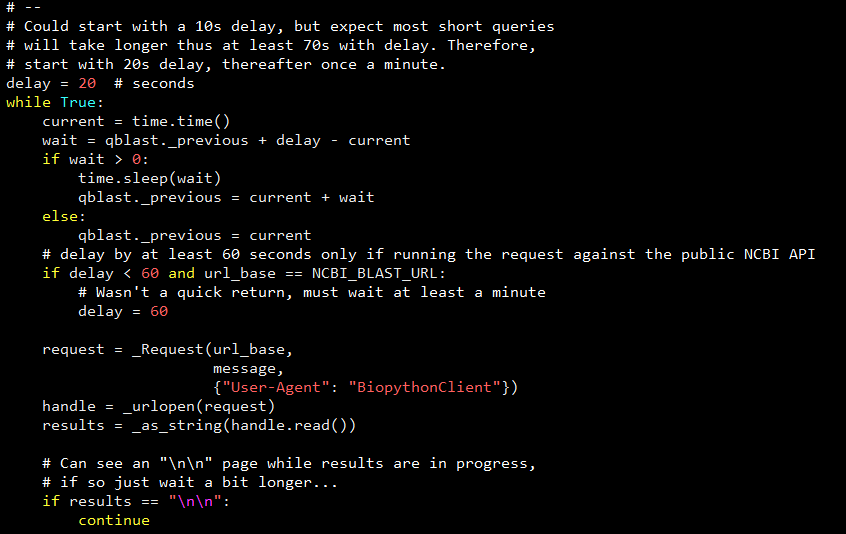
可以看到 NCBIWWW 从 20 秒的延迟开始,然后开始每隔一分钟执行一次 request 轮询,直至任务完成或者任务出现异常。
所以,总的来说,NCBI BLAST API 的使用准则,加上 NCBI BLAST 对用户请求的任务队列处理,甚至 NCBI BLAST 服务器共享资源的限制,以及总用户请求数,这些都可能成为 NCBIWWW.qblast() 异常耗时的原因,这其中还不算个人服务器的网络影响。综上种种原因,如果考虑使用 NCBIWWW.qblast() 执行频繁的序列在线批处理,或许不是一个好的解决方案。
最后,基于 Python 的 NCBI BLAST 在线批处理,如果你有更好的方法,欢迎留言交流。

本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号