预测 motif 的计算原理
本文章来源于简书,作者小潤澤,已获原作者授权;部分内容有调整。
前言
蛋白质中功能的基本单元是 domain,是一种特殊的三维结构,不同结构的 domain 与其他分子特异性结合从而发挥功能。与此类似,转录因子在于 DNA 序列结合时,其结合位点的序列也由于一定的特异性,不同转录因子结合的 DNA 序列的模式是不同的。为了更好的描述结合位点序列的模式,科学家们提出了 motif 的概念。

通常我们对结合位点采用统计每个 site 碱基出现频数。
PFM 矩阵
我们可以登录 JASPAR 网站下载相关的 raw PFM 矩阵(PFM 全称为 position frequency matrix,用于代表 motif 的碱基分布频数)。JASPAR 提供了转录因子与 DNA 结合位点 motif 最全面的公开数据,共收集了脊椎动物、植物、昆虫、线虫、真菌和尾索动物六大类不同类生物的数据。我们随机挑选某一物种的某一转录因子来说明,下载好如同这样:

A,
C,
G,
T 4 种碱基在每个位置的频数分布。
PWM 矩阵
PWM 矩阵在不同文章中有不同的叫法,以下 3 种矩阵其实都是 PWM 矩阵:
position weight matrix (PWM)
position-specific weight matirx (PSWM)
position-specific scoring matrix (PSSM)
在 PFM 矩阵的基础上:
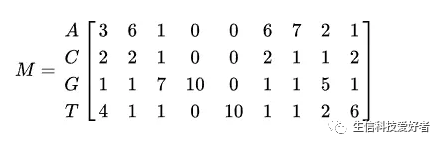
 PPM 矩阵第一行第一列元素为:
PFM 矩阵第一行第一列元素 3/10 = 0.3。
PPM 矩阵第一行第一列元素为:
PFM 矩阵第一行第一列元素 3/10 = 0.3。
GAGGTAAAC
的概率为:
在 PPM 矩阵的基础上做校正就可以得到我们的 PWM 矩阵了。公式为:

即将 PPM 矩阵的每个元素除以背景 0.25(假设基因组上的 ATCG 含量相同,均为 0.25),然后再取 log2。例如, PPM 矩阵第一行第一列的 0.3,log2(0.3/0.25) = 0.26,这样我们就得到了 PWM 矩阵:

GAGGTAAAC 在 PWM 的相应得分为:-1.32 + 1.26 + 1.49 + 2.0 + 2.0+ 1.26 + 1.49 - 0.32 - 1.32 = 6.54。
分数大于 0,说明可能是一个潜在的功能位点,小于 0 则说明是随机序列;你可以根据已有的转录因子结合序列信息(即你感兴趣的转录因子)去预测你待研究的序列是否是这个转录因子的功能位点(根据打分来判断)。
利用 seqLogo 可视化
seqLogo 是一个 R 包,我们读入刚才下载的表格(按 ACGT 的顺序添加表的行名):
data <- read.table("MA0007.2.pfm")
#转换为PPM矩阵
ppm <- sapply(1:ncol(data), function(t){ data[[t]] / sum(data[[t]]) })
#转换为PWM矩阵
p <- makePWM(ppm)
#绘图
seqLogo(p)

Chip-seq 的 motif 分析
在 Chip-seq call peak 中,我们会得到一个 summit.bed,这个其实就是 peak 的峰的碱基,那么只有一个,我们做 motif 分析,只有一个碱基肯定是做不了的。
通常我们用 bedtools slot 来对这个 summit.bed 文件进行 extend,即对这一个峰的碱基前后进行 extend,从而得到一小段序列(extend 100bp),那么就可以做 motif 分析了。


 戳原文,更有料!
戳原文,更有料!
本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号