生信分析流程构建的几大流派
导言
构建生信分析流程是生物信息学从业人员必备的技能之一,对该项能力的评估常常是各大公司招录人员的参考项目之一。
在进行 ngsjs 项目时,我做了一张示意图来表示一些高通量测序数据分析项目重现性的要点(图一)。
ngsjs: A set of command line tools, NGS data analysis workflows, and R shiny plugins/R markdown document for exploring next-generation sequencing data.
一个好的生物信息分析流程可以让你事倍功半,有效减负,同时也有利于他人重复你的数据分析结果。

图一 高通量测序数据分析项目重现性的要点
其中,使用统一的管道(pipeline)、工作流程(workflow)就是其中最重要的一环。
根据生信信息学数据分析流程(管道、工作流程序)构建的风格和方式,大致有以下几大流派(注1):
脚本语言流
Common Workflow language 语言流
Makefile流
配置文件流
Jupyter notebook和R markdown流
......
注1: 围棋比赛中常用“流派”来形容各具特色的棋手/派系,参见资料,深度学习派代表 AlphaGo 现在在围棋届已经一骑绝尘。
生信分析流程构建的几大流派
| 脚本语言流
脚本语言流的主要是通过简单的脚本语言(如 shell,R,Python,Perl)运行各类命令行脚本/程序。常见的几种工作模式:
单个脚本就是一整个流程;
多个脚本组成一个流程;
封装成可以输入参数的命令行程序;
封装成函数/模块/包(包含示例文件、文档和测试)。
前两种(1 和 2)是大多数生物信息学初学者(不具备封装和打包能力)最早开始接触生信分析流程的方式。后两种(3 和 4)是专业人员开发新工具、新流程的必备技能。
这里使用 htslib.org 所给WGS/WES Mapping to Variant Calls (v1.0)作为工作模式1,2的示例(已略去注释):
# mapping
bwa index <ref.fa>
bwa mem -R '@RG\tID:foo\tSM:bar\tLB:library1' <ref.fa> <read1.fa> <read1.fa> > lane.sam
samtools fixmate -O bam <lane.sam> <lane_fixmate.bam>
samtools sort -O bam -o <lane_sorted.bam> -T </tmp/lane_temp> <lane_fixmate.sam>
# Improvement
java -Xmx2g -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -R <ref.fa> -I <lane.bam> -o <lane.intervals> --known <bundle/b38/Mills1000G.b38.vcf>
java -Xmx4g -jar GenomeAnalysisTK.jar -T IndelRealigner -R <ref.fa> -I <lane.bam> -targetIntervals <lane.intervals> --known <bundle/b38/Mills1000G.b38.vcf> -o <lane_realigned.bam>
java -Xmx4g -jar GenomeAnalysisTK.jar -T BaseRecalibrator -R <ref.fa> -knownSites >bundle/b38/dbsnp_142.b38.vcf> -I <lane.bam> -o <lane_recal.table>
java -Xmx2g -jar GenomeAnalysisTK.jar -T PrintReads -R <ref.fa> -I <lane.bam> --BSQR <lane_recal.table> -o <lane_recal.bam>
java -Xmx2g -jar MarkDuplicates.jar VALIDATION_STRINGENCY=LENIENT INPUT=<lane_1.bam> INPUT=<lane_2.bam> INPUT=<lane_3.bam> OUTPUT=<library.bam>
samtools merge <sample.bam> <library1.bam> <library2.bam> <library3.bam>
samtools index <sample.bam>
java -Xmx2g -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -R <ref.fa> -I <sample.bam> -o <sample.intervals> --known >bundle/b38/Mills1000G.b38.vcf>
java -Xmx4g -jar GenomeAnalysisTK.jar -T IndelRealigner -R <ref.fa> -I <sample.bam> -targetIntervals <sample.intervals> --known >bundle/b38/Mills1000G.b38.vcf> -o <sample_realigned.bam>
samtools index <sample_realigned.bam>
# Variant Calling
bcftools mpileup -Ou -f <ref.fa> <sample1.bam> <sample2.bam> <sample3.bam> | bcftools call -vmO z -o <study.vcf.gz>
bcftools mpileup -Ob -o <study.bcf> -f <ref.fa> <sample1.bam> <sample2.bam> <sample3.bam>
bcftools call -vmO z -o <study.vcf.gz> <study.bcf>
tabix -p vcf <study.vcf.gz>
bcftools stats -F <ref.fa> -s - <study.vcf.gz> > <study.vcf.gz.stats>
mkdir plots
plot-vcfstats -p plots/ <study.vcf.gz.stats>
bcftools filter -O z -o <study_filtered..vcf.gz> -s LOWQUAL -i'%QUAL>10' <study.vcf.gz>工作模式3和4是开发生物信息工具的标准方式。很多生物信息学分析工具本身就是涉及到各类复杂计算和命令的集成式流程,如:
deepvariant:A universal SNP and small-indel variant caller using deep neural networks.Nat Biotechnol. 2018 Nov;36(10):983-987. doi: 10.1038/nbt.4235. Epub 2018 Sep 24.
DeepSVR:A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat Genet. 2018 Nov 5. doi: 10.1038/s41588-018-0257-y.
diseasequest:An integrative tissue-network approach to identify and test human disease genes. Nat Biotechnol. 2018 Oct 22. doi: 10.1038/nbt.4246.
bcbio-nextgen:Validated, scalable, community developed variant calling, RNA-seq and small RNA analysis.
chipseq_pipeline:The AQUAS pipeline implements the ENCODE (phase-3) transcription factor and histone ChIP-seq pipeline specifications (by Anshul Kundaje).
deepTools:Tools to process and analyze deep sequencing data. Nucleic Acids Research. 2016 Apr 13:gkw257.
......
使用和开发这类工具/流程的主要原因:
只需要掌握原生编程语言的语法和命令行工具的用法就可以开始构建工具/流程;
其他流程化语言/框架也可以直接调用这些脚本/函数/模块/包/命令行程序;
封装和打包可以减少代码的冗余程度、降低维护难度;
通过使用各类编程语言自带的包管理器解决依赖问题,便于其他用户安装和调用。
我目前主要是 R 语言、Python 写命令行程序、函数、R 包/模块,同时用 CRAN、PyPI 以及 GitHub 分发。
同时,因为 R 语言目前还没有提供一个原生机制直接部署命令行可执行程序(Python、Node包均提供),我现在做了两手准备:
在 ngstkR 包中增加
rbin函数、以及 ngsjs 增加rbin命令行程序一键收集 R 包中inst/bin下面的文件。以 npm 包的形式开发相应的 R 命令行程序,参见正在开发中的 ngsjs 包,初期目标是开发、收集 200+ 和数据分析相关的命令行程序。
目前的体验来看,JavaScript 提供的 npm 和 yarn 包管理工具速度非常快和方便,很适合 R 语言用户同时使用(只需要会写一个 package.json 文件即可)。
| Common Workflow language(CWL)语言
Common Workflow language 语言流是近几年兴起的,专门用于数据分析流程构建的一类语言/工具。
这类语言/工具最核心的部分:定义每一个计算过程(脚本)的输入和输出,然后通过连接这些输入和输出,构成数据分析流程(图二,图三)(如 Galaxy, wdl,cromwell,nextflow,snakemake,bpipe 等)。
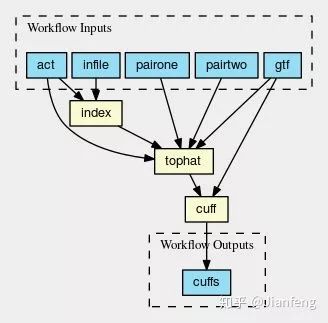
图二 CWL流程示意图一
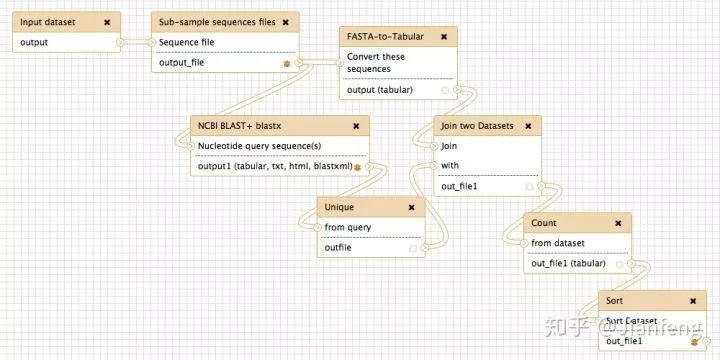
图三 CWL 流程示意图二
示例项目:
chip-seq-pipeline2:ENCODE Transcription Factor and Histone ChIP-Seq processing pipeline (chipseq_pipeline的WDL版).
ngsjs-wkfl-wdl: A library of next-generation sequencing data analysis workflow (WDL).
biowdl: BioWDL is a collection of pipelines and workflows usable for a variety of sequencing related analyses. They are made using WDL and closely related to BIOPET.
snakemake-workflows: An experimental repo for common snakemake rules and workflows.
......
使用和开发这类工具的主要原因:
程序每一步的输入输出参数一目了然;
有图形化流程设计器的支持;
自带日志和运行状态监控功能;
......
扩展阅读:
A review of bioinformatic pipeline frameworks. Brief Bioinform. 2017 May 1;18(3):530-536. doi: 10.1093/bib/bbw020.
Data Harmonization for a Molecularly Driven. Health System. Cell. 2018 Aug 23;174(5):1045-1048. doi: 10.1016/j.cell.2018.08.012.
| Makefile 流
Makefile 流主要是基于软件开发工具 Makefile 的 rule、target 语法运行流程。在 snakemake 工具出现之后(使得数据分析流程支持 CWL),使用Makefile式 Rule 文件构建生物信息学分析流程的用户迅速增加。
pyflow-ATACseq 项目提供的 ATAC-seq 数据分析流程:
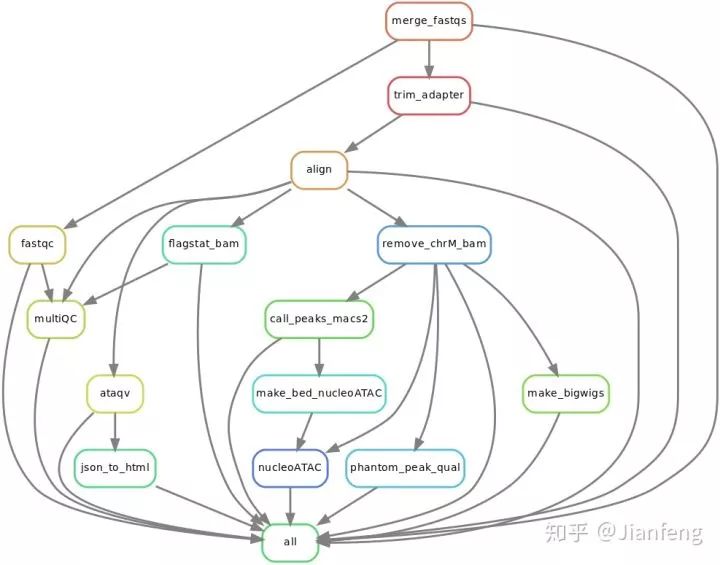
图五 ATAC-seq Snakemake 示例流程图
snakemake 示例文件:
rule targets:
input:
"plots/dataset1.pdf",
"plots/dataset2.pdf"
rule plot:
input:
"raw/{dataset}.csv"
output:
"plots/{dataset}.pdf"
shell:
"somecommand {input} {output}"| 配置文件流
配置文件流(和 CWL 不冲突)主要是基于 JSON、YAML、TOML 等类型的配置文件,然后开发相应的解析器解析和执行流程。如 Galaxy、华为公司最近开源的 Kubegene(基于谷歌开发并开源的容器调度技术 kubernetes)、bashful 的流程文件。
很多计算机软件自动测试流程和构建工具也主要基于配置文件来构建和执行:如 circleci、travis。
这里给出一个基于配置文件的工具示例(图六):
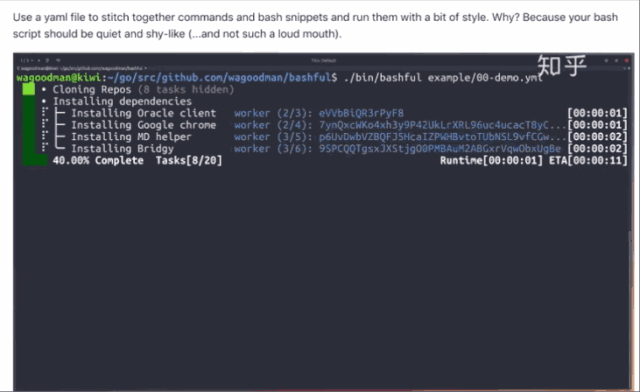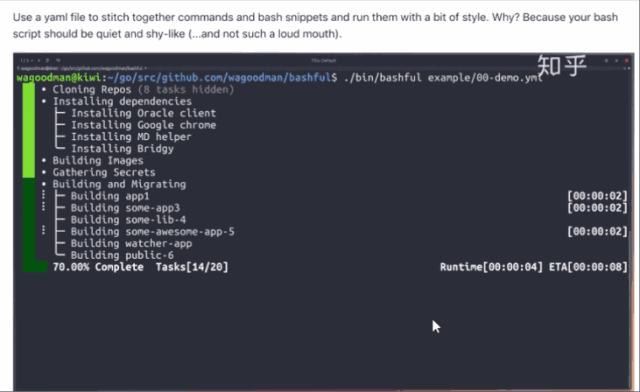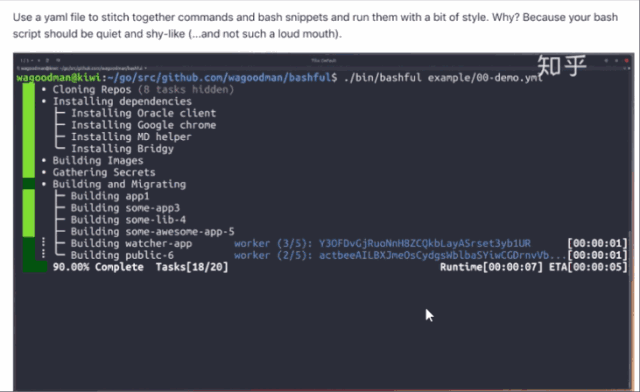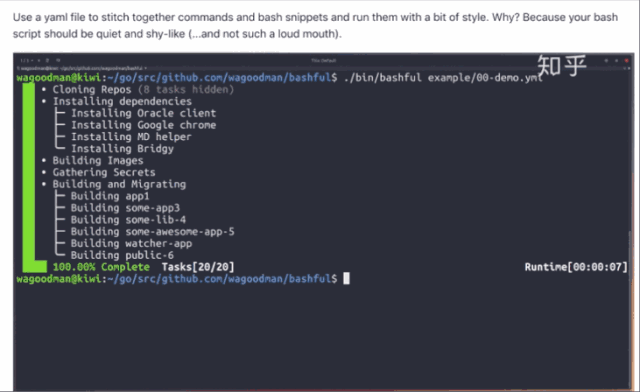
图六 bashful 执行输出
bashful 输入文件格式及部分字段:
config:
show-failure-report: false
show-summary-errors: true
max-parallel-commands: 6
show-task-times: true
x-reference-data:
all-apps: &app-names
- some-lib-4
- utilities-lib
- important-lib
- some-app1
- some-app3
tasks:
- name: Cloning Repos
parallel-tasks:
- name: "Cloning <replace>"
cmd: example/scripts/random-worker.sh 2 <replace>
ignore-failure: true
for-each: *app-names
- name: Building Repos
parallel-tasks:
- name: "Building <replace>"
cmd: example/scripts/random-worker.sh 1 <replace>
ignore-failure: true
for-each: *app-names复杂度较高的生物信息学流程/工具一般至少会提供一个配置文件来管理参数。用户目前也大多接受使用配置文件统一管理变量。
命令行参数也常常结合配置文件同时使用,这么做的主要原因:
可以有效减少动态更新和管理配置文件的次数;
通过命令行修改参数也更加透明和便于日志记录。
|Jupyter notebook 和 R markdown 流
Jupyter notebook 和 R markdown 分别由 Python 语言和 R 语言社区贡献,均可以用于整合文档、代码、以及代码的输出,构建动态、交互式文档和报告系统。
这两个工具已经风靡全世界的数据科学社区,同时也占据了生物信息分析流程中的下游统计分析、建模、以及可视化。
这两个工具兴起的主要原因:
机器学习、高通量测序数据等数据科学的兴起;
大量机器学习、生物信息学分析项目经常需要同时查看文档、即时查看输出、调试代码、进行可视化、撰写报告等;
高质量可视化视图的兴起(颜值的时代);
已有工具使用起来有诸多不便之处,使用门槛相对较高。
Jupyter notebook 示例:
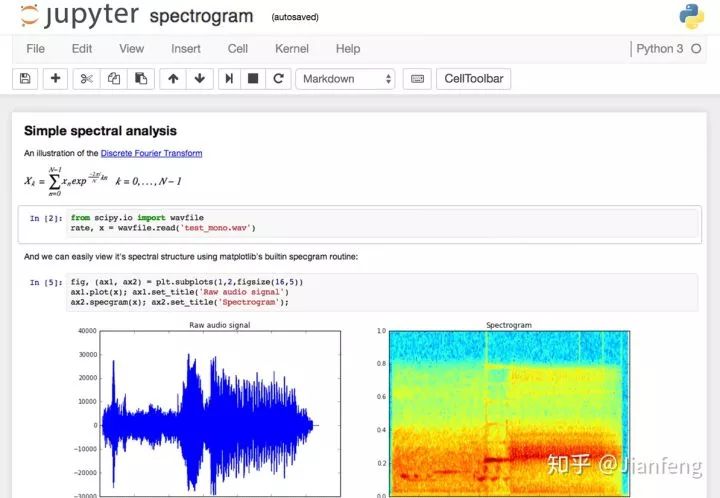
图七 Jupyter notebook
R markdown 示例:
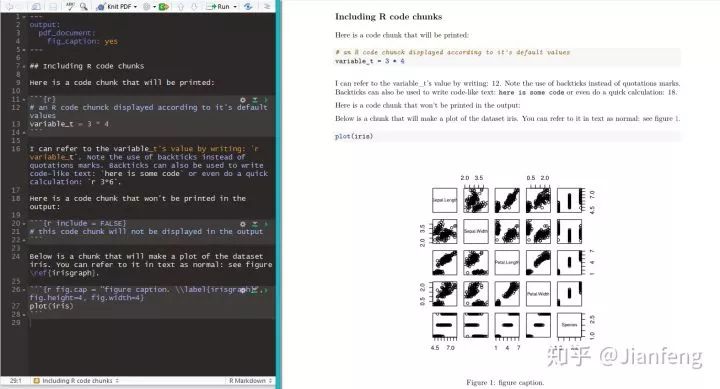
图八 Jupyter notebook
以 R 语言为例,在一个 R 包开发过程中,常常集成 R markdown 文件来动态更新文档、教程和项目主页。
比如其中我开发的两个项目 configr、BioInstaller:
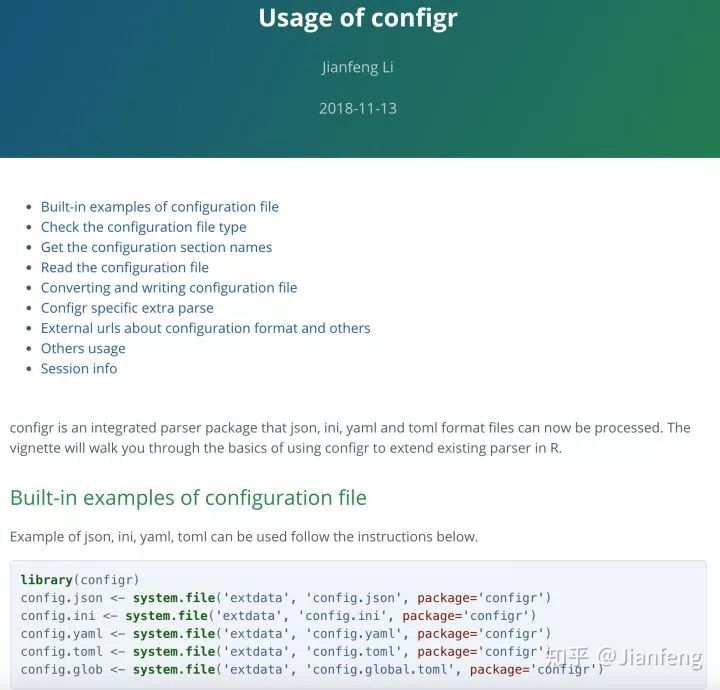
图九 configr 说明文档
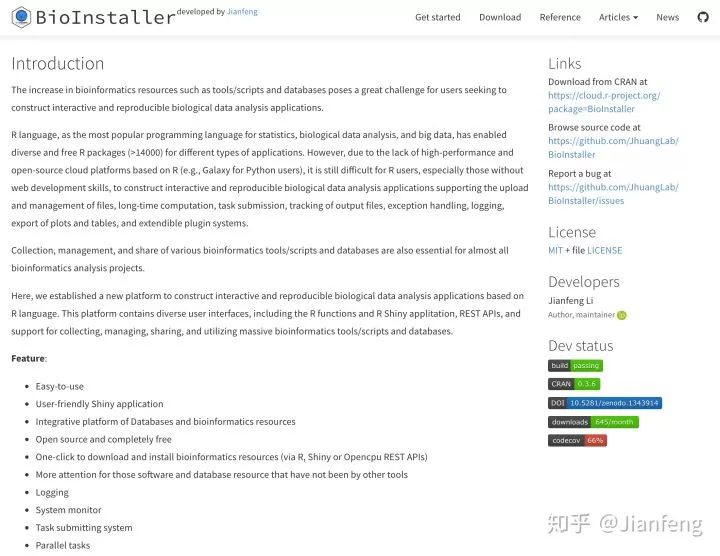
图十 BioInstaller 项目主页
相关的 R 包:
blogdown:辅助个人博客创建和维护;
bookdown:辅助数据科学书籍的构建;
xaringan:辅助创作 Web PPT;
pkgdown:一键生成 R 项目主页;
shiny:辅助 R markdown 构建更复杂的交互式文档;
future:简化 R 语言用户的并行化操作。
我在这里设想了一个 R markdown 的应用场景:
用户使用 R markdown 并通过连接数据库、访问网页 APIs 的方式提交数据分析任务;
构建文档即重新检查数据分析进程和状态、生成相应的运行状态可视化、表格等监控结果;
完成上游数据分析之后可以直接开始进行下游个性化的数据统计分析和可视化、同时撰写结题报告;
同时使用 Shiny 应用/其他通过网页服务展示分析结果。
| 其他
软件和科学社区一直会有新的工具、思想、范式出现,生物信息学数据分析流程也不例外,我在这篇文章中所列的几种方式只能大致涵盖目前比较主流的几种方式。
还有一些”非主流“流程构建方式:
博导流: “A 同学你过来一下,我们讨论一下你的课题,你可能需要写一个 Pipeline,输入病人 DNA、RNA 的测序 fastq 文件、表型数据,输出所有可以完成的生物信息数据分析结果”
AI 流: “你好,Siri,帮我写一个 Pipeline,输入...;输出....”
公式流
output (multi_omics) ~ input(2 * DNA@wgs@fq + 2 * DNA@wes@fq + DNA@chip_seq + 2 * RNA@rnaseq@fq + n * RNA@scrna + ...)
output (paper) ~ input(patient@survive + patient@habits_and_customs + mutation@proteinpaint + —
END—
—
END—

本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号