深入浅出介绍聚类分析

点击蓝字获取更多精彩信息
聚类分析是生信分析中常用的工具,在转录组分析中经常用到。聚类分析将表达模式相似的基因聚类在一起,以基因集的形式进行后续分析,今天小编给大家介绍其相关原理。
介绍
聚类方法有很多,常用的有以下几个:
k-均值聚类(k-means Cluster)
层次聚类(Hierarchical Cluster)
SOM(自组织映射)
FCM(模糊 C 均值)
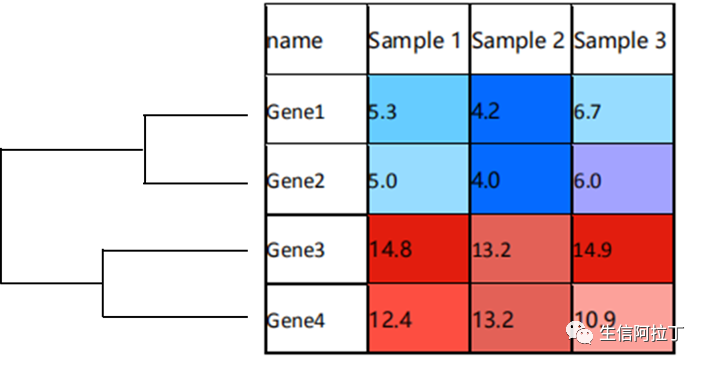
下图的例子展示的是,差异表达基因集的聚类热图。
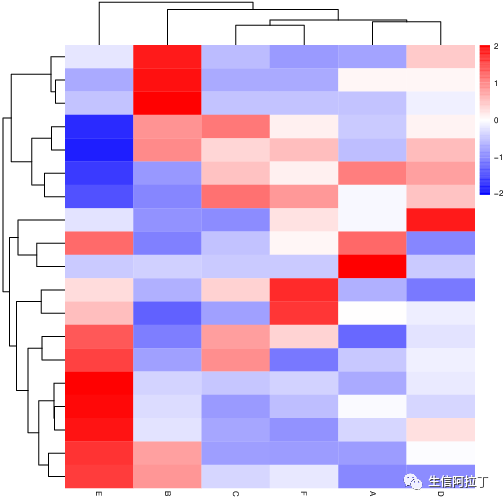
多是基于 R 语言 heatmap.2 函数绘制(gplots 程序包),该函数默认使用的聚类方法是计算欧式距离(Euclidean Distance)进行层次聚类(Hierarchical Cluster)。
这个图的是什么意思呢?我们来解释一下。
每个小方格表示一个基因,颜色则表示该基因的表达量;
每一行表示同一个基因在不同样本的表达情况;
每列表示一个样本中不同基因的表达情况;
上方的聚类是表示对来自不同样本的聚类结果;
左侧的树状图是表示对来自不同样本的不同基因的聚类分析结果。
什么是距离?
首先,我们先明确下什么是欧式距离(Euclidean Distance):
欧式距离,也称欧几里得距离,是衡量多维空间的两个点之间的绝对距离,
(1) 二维平面,两点 a(x1,y1),b(x2,y2) 欧式距离的计算公式为:
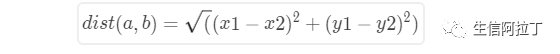
(2) 三维空间,欧式距离的计算公式为:
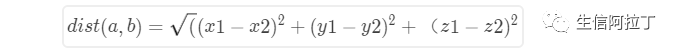
(3) n维空间,欧式距离的计算公式为:
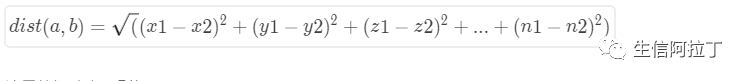
那么,体现在基因表达量的矩阵上,则如下:
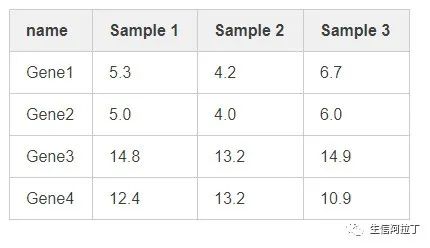
(1) 首行为样本名;
(2) 首列为基因名;
(3) 数字则为基因在相应样本中的表达量(一般使用标准化后的表达量矩阵)
Gene1 与 Gene2 的欧式距离为:
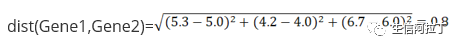
Gene1 与 Gene3 的欧式距离为:
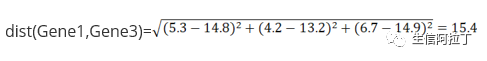
Gene1 与 Gene4 的欧式距离为:
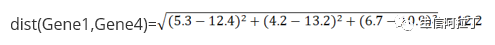
聚类的过程
计算出所有基因两两之间的欧式距离之后,就可以进行聚类啦:
找出欧式距离最近的两个基因首先形成一簇 Cluster1。由于,Gene1 与 Gene2 的欧式距离要小于 Gene1 与 Gene3,Gene4 的欧式距离,Gene1 与 Gene2 会形成一簇 Cluster1;
将 Cluster1 作为一个整体,计算其与其他基因两两之间的欧式距离,并且寻找欧式距离最近的两个基因(或者 cluster)形成新的 Cluster2。例如,可算得 Gene3 和 Gene4 的欧式聚类也小于同其他基因的欧式距离,Gene3 和 Gene4 也会形成一簇 Cluster2;
如此反复,直到所有的聚类完成。
Cluster 之间的聚类,则有3种方法:
重心法(centroid)
最短距离法(single-linkage)
最长距离法(complete-linkage)
R 语言中 hclust 函数的默认方法为最长距离法(complete-linkage)。
以上的聚类过程即称之为层级聚类。
层级聚类一般伴随着系统聚类图,系统聚类图分支的长短也体现 Cluster 形成的早晚,分支越短,形成的越早,基因表达模式也越相近。
总结
聚类分析将基因划分为不同的基因集合,用于反映不同实验条件下样品差异表达基因的变化模式。
功能相关的基因在相同条件下通常具有相似的表达模式,例如被共同的转录因子调控的基因,或其产物构成同一个蛋白复合体的基因,或参与相同生物学过程的基因。对这些基因集进行分析往往可以获得比单基因分析更为可靠的结果。
获得基因集之后,可以进行通路分析、富集分析,以及更高级的 GSEA 或者 WGCNA 分析,大家请继续关注我们后续吧。

作者:麦茬道
审稿:童蒙
编辑:amethyst

本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。

