RNA-seq分析全流程
安装conda
wget -c https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh# 取消conda自动激活
conda config --set auto_activate_base false#创建conda环境
conda create -n RNA-seq python=3.6.2# 添加几个通道
conda config --add channels r
conda config --add channels defaults
conda config --add channels conda-forge
conda config --add channels bioconda
# 无论是conda默认的软件源还是bioconda软件源都是国外的,速度非常慢,所以需要增加国内软件源,同时bioconda已经有清华,中科大两个国内镜像,也添加进去
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --set show_channel_urls yes ## 设置搜索时显示通道地址
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/msys2/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
#查看目前conda软件源情况
conda info
下载水稻的参考基因组文件和注释文件
wget -c https://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_genome.fasta.gz # 下载基因组文件
wget -c https://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_representative_2021-05-10.tar.gz # 下载注释文件
下载水稻冷胁迫下测序数据
# 安装sratoolkit
conda install -c bioconda sra-tools
# 下载数据
cat SRR_Acc_List.txt | xargs prefetch -v将sra文件转换为fq文件
for id in `seq 10 25`;
do
fastq-dump --gzip --split-3 -O ~/data/RNA-Seq/fastq -A SRR66558${id}/*
done
# 该步骤耗时较长,可并行得到了fastq文件我们就可以采用不同的RNA-seq protocol来进行分析
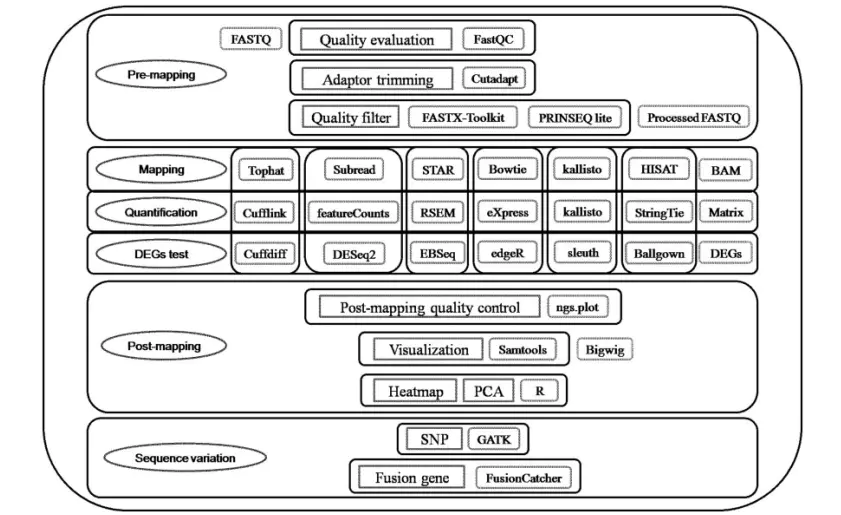
本应该有数据质量检测,此处略过
建立基因组序列索引
# 安装hisat2
conda install hista2
# 建立存放索引文件的目录
mkdir rice_hisat2_index
hisat2-build -p 16 IRGSP-1.0_genome.fasta rice
# 建立存放水稻注释文件的目录
mkdir rice_gff将测序数据比对到参考基因组上
for i in `seq 10 25`
do
hisat2 -p 16 -x /home/hgdai/RNA-seq/ref/rice_hista2_index/rice -1 /home/hgdai/RNA-seq/fastq/SRR66558${i}_1.fastq.gz -2 /home/hgdai/RNA-seq/fastq/SRR66558${i}_2.fastq.gz -S /home/hgdai/RNA-seq/align/SRR66558${i}.sam --new-summary --summary-file /home/hgdai/RNA-seq/fastq/SRR66558${i}.ht2.txt
done
