
Feature Selection in Machine Learning (Breast Cancer Datasets)
Machine learning uses so called features (i.e. variables or attributes) to generate predictive models. Using a suitable combination of features is essential for obtaining high precision and accuracy. Because too many (unspecific) features pose the problem of overfitting the model, we generally want to restrict the features in our models to those, that are most relevant for the response variable we want to predict. Using as few features as possible will also reduce the complexity of our models, which means it needs less time and computer power to run and is easier to understand.
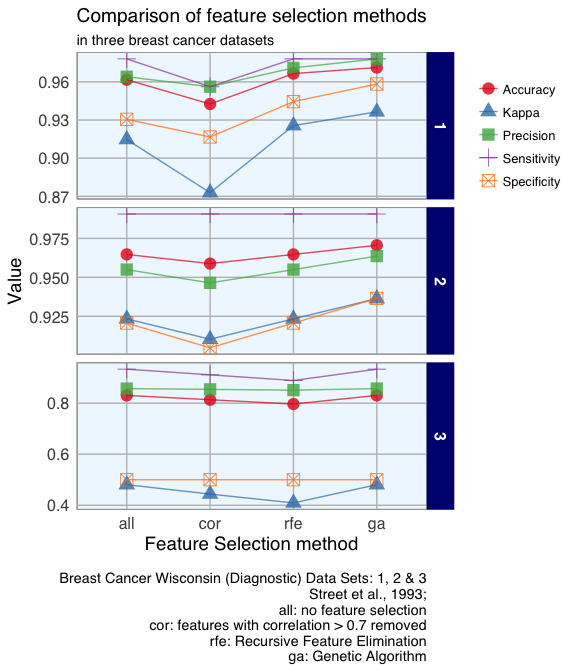
There are several ways to identify how much each feature contributes to the model and to restrict the number of selected features. Here, I am going to examine the effect of feature selection via
- Correlation,
- Recursive Feature Elimination (RFE) and
- Genetic Algorithm (GA)
on Random Forest models.
Additionally, I want to know how different data properties affect the influence of these feature selection methods on the outcome. For that I am using three breast cancer datasets, one of which has few features; the other two are larger but differ in how well the outcome clusters in PCA.
Based on my comparisons of the correlation method, RFE and GA, I would conclude that for Random Forest models
- removing highly correlated features isn’t a generally suitable method,
- GA produced the best models in this example but is impractical for everyday use-cases with many features because it takes a lot of time to run with sufficient generations and individuals and
- data that doesn’t allow a good classification to begin with (because the features are not very distinct between classes) don’t necessarily benefit from feature selection.
My conclusions are of course not to be generalized to any ol’ data you are working with: There are many more feature selection methods and I am only looking at a limited number of datasets and only at their influence on Random Forest models. But even this small example shows how different features and parameters can influence your predictions. With machine learning, there is no “one size fits all”! It is always worthwhile to take a good hard look at your data, get acquainted with its quirks and properties before you even think about models and algorithms. And once you’ve got a feel for your data, investing the time and effort to compare different feature selection methods (or engineered features), model parameters and - finally - different machine learning algorithms can make a big difference!
Breast Cancer Wisconsin (Diagnostic) Dataset
The data I am going to use to explore feature selection methods is the Breast Cancer Wisconsin (Diagnostic) Dataset:
W.N. Street, W.H. Wolberg and O.L. Mangasarian. Nuclear feature extraction for breast tumor diagnosis. IS&T/SPIE 1993 International Symposium on Electronic Imaging: Science and Technology, volume 1905, pages 861-870, San Jose, CA, 1993.
O.L. Mangasarian, W.N. Street and W.H. Wolberg. Breast cancer diagnosis and prognosis via linear programming. Operations Research, 43(4), pages 570-577, July-August 1995.
W.H. Wolberg, W.N. Street, and O.L. Mangasarian. Machine learning techniques to diagnose breast cancer from fine-needle aspirates. Cancer Letters 77 (1994) 163-171.
W.H. Wolberg, W.N. Street, and O.L. Mangasarian. Image analysis and machine learning applied to breast cancer diagnosis and prognosis. Analytical and Quantitative Cytology and Histology, Vol. 17 No. 2, pages 77-87, April 1995.
W.H. Wolberg, W.N. Street, D.M. Heisey, and O.L. Mangasarian. Computerized breast cancer diagnosis and prognosis from fine needle aspirates. Archives of Surgery 1995;130:511-516.
W.H. Wolberg, W.N. Street, D.M. Heisey, and O.L. Mangasarian. Computer-derived nuclear features distinguish malignant from benign breast cytology. Human Pathology, 26:792–796, 1995.
The data was downloaded from the UC Irvine Machine Learning Repository. The features in these datasets characterise cell nucleus properties and were generated from image analysis of fine needle aspirates (FNA) of breast masses.
Included are three datasets. The first dataset is small with only 9 features, the other two datasets have 30 and 33 features and vary in how strongly the two predictor classes cluster in PCA. I want to explore the effect of different feature selection methods on datasets with these different properties.
But first, I want to get to know the data I am working with.
Breast cancer dataset 1
The first dataset looks at the predictor classes:
- malignant or
- benign breast mass.
The phenotypes for characterisation are:
- Sample ID (code number)
- Clump thickness
- Uniformity of cell size
- Uniformity of cell shape
- Marginal adhesion
- Single epithelial cell size
- Number of bare nuclei
- Bland chromatin
- Number of normal nuclei
- Mitosis
- Classes, i.e. diagnosis
Missing values are imputed with the mice package.
bc_data <- read.table("breast-cancer-wisconsin.data.txt", header = FALSE, sep = ",")
colnames(bc_data) <- c("sample_code_number", "clump_thickness", "uniformity_of_cell_size", "uniformity_of_cell_shape", "marginal_adhesion", "single_epithelial_cell_size",
"bare_nuclei", "bland_chromatin", "normal_nucleoli", "mitosis", "classes")
bc_data$classes <- ifelse(bc_data$classes == "2", "benign",
ifelse(bc_data$classes == "4", "malignant", NA))
bc_data[bc_data == "?"] <- NA
# how many NAs are in the data
length(which(is.na(bc_data)))## [1] 16
# impute missing data
library(mice)
bc_data[,2:10] <- apply(bc_data[, 2:10], 2, function(x) as.numeric(as.character(x)))
dataset_impute <- mice(bc_data[, 2:10], print = FALSE)
bc_data <- cbind(bc_data[, 11, drop = FALSE], mice::complete(dataset_impute, 1))
bc_data$classes <- as.factor(bc_data$classes)
# how many benign and malignant cases are there?
summary(bc_data$classes)## benign malignant
## 458 241
str(bc_data)## 'data.frame': 699 obs. of 10 variables:
## $ classes : Factor w/ 2 levels "benign","malignant": 1 1 1 1 1 2 1 1 1 1 ...
## $ clump_thickness : num 5 5 3 6 4 8 1 2 2 4 ...
## $ uniformity_of_cell_size : num 1 4 1 8 1 10 1 1 1 2 ...
## $ uniformity_of_cell_shape : num 1 4 1 8 1 10 1 2 1 1 ...
## $ marginal_adhesion : num 1 5 1 1 3 8 1 1 1 1 ...
## $ single_epithelial_cell_size: num 2 7 2 3 2 7 2 2 2 2 ...
## $ bare_nuclei : num 1 10 2 4 1 10 10 1 1 1 ...
## $ bland_chromatin : num 3 3 3 3 3 9 3 3 1 2 ...
## $ normal_nucleoli : num 1 2 1 7 1 7 1 1 1 1 ...
## $ mitosis : num 1 1 1 1 1 1 1 1 5 1 ...
Breast cancer dataset 2
The second dataset looks again at the predictor classes:
- M: malignant or
- B: benign breast mass.
The first two columns give:
- Sample ID
- Classes, i.e. diagnosis
For each cell nucleus, the following ten characteristics were measured:
- Radius (mean of all distances from the center to points on the perimeter)
- Texture (standard deviation of gray-scale values)
- Perimeter
- Area
- Smoothness (local variation in radius lengths)
- Compactness (perimeter^2 / area - 1.0)
- Concavity (severity of concave portions of the contour)
- Concave points (number of concave portions of the contour)
- Symmetry
- Fractal dimension (“coastline approximation” - 1)
For each characteristic three measures are given:
- Mean
- Standard error
- Largest/ “worst”
bc_data_2 <- read.table("wdbc.data.txt", header = FALSE, sep = ",")
phenotypes <- rep(c("radius", "texture", "perimeter", "area", "smoothness", "compactness", "concavity", "concave_points", "symmetry", "fractal_dimension"), 3)
types <- rep(c("mean", "se", "largest_worst"), each = 10)
colnames(bc_data_2) <- c("ID", "diagnosis", paste(phenotypes, types, sep = "_"))
# how many NAs are in the data
length(which(is.na(bc_data_2)))## [1] 0
# how many benign and malignant cases are there?
summary(bc_data_2$diagnosis)## B M
## 357 212
str(bc_data_2)## 'data.frame': 569 obs. of 32 variables:
## $ ID : int 842302 842517 84300903 84348301 84358402 843786 844359 84458202 844981 84501001 ...
## $ diagnosis : Factor w/ 2 levels "B","M": 2 2 2 2 2 2 2 2 2 2 ...
## $ radius_mean : num 18 20.6 19.7 11.4 20.3 ...
## $ texture_mean : num 10.4 17.8 21.2 20.4 14.3 ...
## $ perimeter_mean : num 122.8 132.9 130 77.6 135.1 ...
## $ area_mean : num 1001 1326 1203 386 1297 ...
## $ smoothness_mean : num 0.1184 0.0847 0.1096 0.1425 0.1003 ...
## $ compactness_mean : num 0.2776 0.0786 0.1599 0.2839 0.1328 ...
## $ concavity_mean : num 0.3001 0.0869 0.1974 0.2414 0.198 ...
## $ concave_points_mean : num 0.1471 0.0702 0.1279 0.1052 0.1043 ...
## $ symmetry_mean : num 0.242 0.181 0.207 0.26 0.181 ...
## $ fractal_dimension_mean : num 0.0787 0.0567 0.06 0.0974 0.0588 ...
## $ radius_se : num 1.095 0.543 0.746 0.496 0.757 ...
## $ texture_se : num 0.905 0.734 0.787 1.156 0.781 ...
## $ perimeter_se : num 8.59 3.4 4.58 3.44 5.44 ...
## $ area_se : num 153.4 74.1 94 27.2 94.4 ...
## $ smoothness_se : num 0.0064 0.00522 0.00615 0.00911 0.01149 ...
## $ compactness_se : num 0.049 0.0131 0.0401 0.0746 0.0246 ...
## $ concavity_se : num 0.0537 0.0186 0.0383 0.0566 0.0569 ...
## $ concave_points_se : num 0.0159 0.0134 0.0206 0.0187 0.0188 ...
## $ symmetry_se : num 0.03 0.0139 0.0225 0.0596 0.0176 ...
## $ fractal_dimension_se : num 0.00619 0.00353 0.00457 0.00921 0.00511 ...
## $ radius_largest_worst : num 25.4 25 23.6 14.9 22.5 ...
## $ texture_largest_worst : num 17.3 23.4 25.5 26.5 16.7 ...
## $ perimeter_largest_worst : num 184.6 158.8 152.5 98.9 152.2 ...
## $ area_largest_worst : num 2019 1956 1709 568 1575 ...
## $ smoothness_largest_worst : num 0.162 0.124 0.144 0.21 0.137 ...
## $ compactness_largest_worst : num 0.666 0.187 0.424 0.866 0.205 ...
## $ concavity_largest_worst : num 0.712 0.242 0.45 0.687 0.4 ...
## $ concave_points_largest_worst : num 0.265 0.186 0.243 0.258 0.163 ...
## $ symmetry_largest_worst : num 0.46 0.275 0.361 0.664 0.236 ...
## $ fractal_dimension_largest_worst: num 0.1189 0.089 0.0876 0.173 0.0768 ...
Breast cancer dataset 3
The third dataset looks at the predictor classes:
- R: recurring or
- N: nonrecurring breast cancer.
The first two columns give:
- Sample ID
- Classes, i.e. outcome
For each cell nucleus, the same ten characteristics and measures were given as in dataset 2, plus:
- Time (recurrence time if field 2 = R, disease-free time if field 2 = N)
- Tumor size - diameter of the excised tumor in centimeters
- Lymph node status - number of positive axillary lymph nodes observed at time of surgery
Missing values are imputed with the mice package.
bc_data_3 <- read.table("wpbc.data.txt", header = FALSE, sep = ",")
colnames(bc_data_3) <- c("ID", "outcome", "time", paste(phenotypes, types, sep = "_"), "tumor_size", "lymph_node_status")
bc_data_3[bc_data_3 == "?"] <- NA
# how many NAs are in the data
length(which(is.na(bc_data_3)))## [1] 4
# impute missing data
library(mice)
bc_data_3[,3:35] <- apply(bc_data_3[,3:35], 2, function(x) as.numeric(as.character(x)))
dataset_impute <- mice(bc_data_3[,3:35], print = FALSE)
bc_data_3 <- cbind(bc_data_3[, 2, drop = FALSE], mice::complete(dataset_impute, 1))
# how many recurring and non-recurring cases are there?
summary(bc_data_3$outcome)## N R
## 151 47
str(bc_data_3)## 'data.frame': 198 obs. of 34 variables:
## $ outcome : Factor w/ 2 levels "N","R": 1 1 1 1 2 2 1 2 1 1 ...
## $ time : num 31 61 116 123 27 77 60 77 119 76 ...
## $ radius_mean : num 18 18 21.4 11.4 20.3 ...
## $ texture_mean : num 27.6 10.4 17.4 20.4 14.3 ...
## $ perimeter_mean : num 117.5 122.8 137.5 77.6 135.1 ...
## $ area_mean : num 1013 1001 1373 386 1297 ...
## $ smoothness_mean : num 0.0949 0.1184 0.0884 0.1425 0.1003 ...
## $ compactness_mean : num 0.104 0.278 0.119 0.284 0.133 ...
## $ concavity_mean : num 0.109 0.3 0.126 0.241 0.198 ...
## $ concave_points_mean : num 0.0706 0.1471 0.0818 0.1052 0.1043 ...
## $ symmetry_mean : num 0.186 0.242 0.233 0.26 0.181 ...
## $ fractal_dimension_mean : num 0.0633 0.0787 0.0601 0.0974 0.0588 ...
## $ radius_se : num 0.625 1.095 0.585 0.496 0.757 ...
## $ texture_se : num 1.89 0.905 0.611 1.156 0.781 ...
## $ perimeter_se : num 3.97 8.59 3.93 3.44 5.44 ...
## $ area_se : num 71.5 153.4 82.2 27.2 94.4 ...
## $ smoothness_se : num 0.00443 0.0064 0.00617 0.00911 0.01149 ...
## $ compactness_se : num 0.0142 0.049 0.0345 0.0746 0.0246 ...
## $ concavity_se : num 0.0323 0.0537 0.033 0.0566 0.0569 ...
## $ concave_points_se : num 0.00985 0.01587 0.01805 0.01867 0.01885 ...
## $ symmetry_se : num 0.0169 0.03 0.0309 0.0596 0.0176 ...
## $ fractal_dimension_se : num 0.00349 0.00619 0.00504 0.00921 0.00511 ...
## $ radius_largest_worst : num 21.6 25.4 24.9 14.9 22.5 ...
## $ texture_largest_worst : num 37.1 17.3 21 26.5 16.7 ...
## $ perimeter_largest_worst : num 139.7 184.6 159.1 98.9 152.2 ...
## $ area_largest_worst : num 1436 2019 1949 568 1575 ...
## $ smoothness_largest_worst : num 0.119 0.162 0.119 0.21 0.137 ...
## $ compactness_largest_worst : num 0.193 0.666 0.345 0.866 0.205 ...
## $ concavity_largest_worst : num 0.314 0.712 0.341 0.687 0.4 ...
## $ concave_points_largest_worst : num 0.117 0.265 0.203 0.258 0.163 ...
## $ symmetry_largest_worst : num 0.268 0.46 0.433 0.664 0.236 ...
## $ fractal_dimension_largest_worst: num 0.0811 0.1189 0.0907 0.173 0.0768 ...
## $ tumor_size : num 5 3 2.5 2 3.5 2.5 1.5 4 2 6 ...
## $ lymph_node_status : num 5 2 0 0 0 0 0 10 1 20 ...
Principal Component Analysis (PCA)
To get an idea about the dimensionality and variance of the datasets, I am first looking at PCA plots for samples and features. The first two principal components (PCs) show the two components that explain the majority of variation in the data.
After defining my custom ggplot2 theme, I am creating a function that performs the PCA (using the pcaGoPromoter package), calculates ellipses of the data points (with the ellipsepackage) and produces the plot with ggplot2. Some of the features in datasets 2 and 3 are not very distinct and overlap in the PCA plots, therefore I am also plotting hierarchical clustering dendrograms.
# plotting theme
library(ggplot2)
my_theme <- function(base_size = 12, base_family = "sans"){
theme_minimal(base_size = base_size, base_family = base_family) +
theme(
axis.text = element_text(size = 12),
axis.text.x = element_text(angle = 0, vjust = 0.5, hjust = 0.5),
axis.title = element_text(size = 14),
panel.grid.major = element_line(color = "grey"),
panel.grid.minor = element_blank(),
panel.background = element_rect(fill = "aliceblue"),
strip.background = element_rect(fill = "navy", color = "navy", size = 1),
strip.text = element_text(face = "bold", size = 12, color = "white"),
legend.position = "right",
legend.justification = "top",
legend.background = element_blank(),
panel.border = element_rect(color = "grey", fill = NA, size = 0.5)
)
}
theme_set(