


Linux command line exercises for NGS data processing
by Umer Zeeshan Ijaz
The purpose of this tutorial is to introduce students to the frequently used tools for NGS analysis as well as giving experience in writing one-liners. Copy the required files to your current directory, change directory (cd) to thelinuxTutorial folder, and do all the processing inside:
[uzi@quince-srv2 ~/]$ cp -r /home/opt/MScBioinformatics/linuxTutorial .
[uzi@quince-srv2 ~/]$ cd linuxTutorial
[uzi@quince-srv2 ~/linuxTutorial]$
I have deliberately chosen Awk in the exercises as it is a language in itself and is used more often to manipulate NGS data as compared to the other command line tools such as grep, sed, perl etc. Furthermore, having a command on awkwill make it easier to understand advanced tutorials such as Illumina Amplicons Processing Workflow.
In Linux, we use a shell that is a program that takes your commands from the keyboard and gives them to the operating system. Most Linux systems utilize Bourne Again SHell (bash), but there are several additional shell programs on a typical Linux system such as ksh, tcsh, and zsh. To see which shell you are using, type
[uzi@quince-srv2 ~/linuxTutorial]$ echo $SHELL
/bin/bash
To see where you are in the file system:
[uzi@quince-srv2 ~/linuxTutorial]$ pwd
/home/uzi/linuxTutorial
List the files in the current directory:
[uzi@quince-srv2 ~/linuxTutorial]$ ls
data
Now try different commands from the sheet given below:
Linux Commands Cheat Sheet
-
File System
ls— list items in current directoryls -l— list items in current directory and show in long format to see perimissions, size, and modification datels -a— list all items in current directory, including hidden filesls -F— list all items in current directory and show directories with a slash and executables with a starls dir— list all items in directory dircd dir— change directory to dircd ..— go up one directorycd /— go to the root directorycd ~— go to to your home directorycd -— go to the last directory you were just inpwd— show present working directorymkdir dir— make directory dirrm file— remove filerm -r dir— remove directory dir recursivelycp file1 file2— copy file1 to file2cp -r dir1 dir2— copy directory dir1 to dir2 recursivelymv file1 file2— move (rename) file1 to file2ln -s file link— create symbolic link to filetouch file— create or update filecat file— output the contents of fileless file— view file with page navigationhead file— output the first 10 lines of filetail file— output the last 10 lines of filetail -f file— output the contents of file as it grows, starting with the last 10 linesvim file— edit filealias name 'command'— create an alias for a command -
System
shutdown— shut down machinereboot— restart machinedate— show the current date and timewhoami— who you are logged in asfinger user— display information about userman command— show the manual for commanddf— show disk usagedu— show directory space usagefree— show memory and swap usagewhereis app— show possible locations of appwhich app— show which app will be run by default -
Process Management
ps— display your currently active processestop— display all running processeskill pid— kill process id pidkill -9 pid— force kill process id pid -
Permissions
ls -l— list items in current directory and show permissionschmod ugo file— change permissions of file to ugo - u is the user's permissions, g is the group's permissions, and o is everyone else's permissions. The values of u, g, and o can be any number between 0 and 7.7— full permissions6— read and write only5— read and execute only4— read only3— write and execute only2— write only1— execute only0— no permissionschmod 600 file— you can read and write - good for fileschmod 700 file— you can read, write, and execute - good for scriptschmod 644 file— you can read and write, and everyone else can only read - good for web pageschmod 755 file— you can read, write, and execute, and everyone else can read and execute - good for programs that you want to share -
Networking
wget file— download a filecurl file— download a filescp user@host:file dir— secure copy a file from remote server to the dir directory on your machinescp file user@host:dir— secure copy a file from your machine to the dir directory on a remote serverscp -r user@host:dir dir— secure copy the directory dir from remote server to the directory dir on your machinessh user@host— connect to host as userssh -p port user@host— connect to host on port as userssh-copy-id user@host— add your key to host for user to enable a keyed or passwordless loginping host— ping host and output resultswhois domain— get information for domaindig domain— get DNS information for domaindig -x host— reverse lookup hostlsof -i tcp:1337— list all processes running on port 1337 -
Searching
grep pattern files— search for pattern in filesgrep -r pattern dir— search recursively for pattern in dirgrep -rn pattern dir— search recursively for pattern in dir and show the line number foundgrep -r pattern dir --include='*.ext— search recursively for pattern in dir and only search in files with .ext extensioncommand | grep pattern— search for pattern in the output of commandfind file— find all instances of file in real systemlocate file— find all instances of file using indexed database built from the updatedb command. Much faster than findsed -i 's/day/night/g' file— find all occurrences of day in a file and replace them with night - s means substitude and g means global - sed also supports regular expressions -
Compression
tar cf file.tar files— create a tar named file.tar containing filestar xf file.tar— extract the files from file.tartar czf file.tar.gz files— create a tar with Gzip compressiontar xzf file.tar.gz— extract a tar using Gzipgzip file— compresses file and renames it to file.gzgzip -d file.gz— decompresses file.gz back to file -
Shortcuts
ctrl+a— move cursor to beginning of linectrl+f— move cursor to end of linealt+f— move cursor forward 1 wordalt+b— move cursor backward 1 word
http://cheatsheetworld.com/programming/unix-linux-cheat-sheet/Exercise 1: Extracting reads from a FASTA file based on supplied IDs
Awk is a programming language which allows easy manipulation of structured data and is mostly used for pattern scanning and processing. It searches one or more files to see if they contain lines that match with the specified patterns and then perform associated actions. The basic syntax is:
awk '/pattern1/ {Actions}
/pattern2/ {Actions}' file
The working of Awk is as follows
Awkreads the input files one line at a time.- For each line, it matches with given pattern in the given order, if matches performs the corresponding action.
- If no pattern matches, no action will be performed.
- In the above syntax, either search pattern or action are optional, But not both.
- If the search pattern is not given, then
Awkperforms the given actions for each line of the input. - If the action is not given, print all that lines that matches with the given patterns which is the default action.
- Empty braces with out any action does nothing. It wont perform default printing operation.
- Each statement in Actions should be delimited by semicolon.
data.tsv with the following contents:
$ cat data/test.tsv blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG blah_C2 ACTTTATATATT blah_C3 ACTTATATATATATA blah_C4 ACTTATATATATATA blah_C5 ACTTTATATATTBy default
Awk prints every line from the file.
$ awk '{print;}' data/test.tsv
blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
blah_C2 ACTTTATATATT
blah_C3 ACTTATATATATATA
blah_C4 ACTTATATATATATA
blah_C5 ACTTTATATATT
We print the line which matches the pattern blah_C3
$ awk '/blah_C3/' data/test.tsv blah_C3 ACTTATATATATATA
Awk has number of builtin variables. For each record i.e line, it splits the record delimited by whitespace character by default and stores it in the $n variables. If the line has 5 words, it will be stored in $1, $2, $3, $4 and $5.$0 represents the whole line. NF is a builtin variable which represents the total number of fields in a record.
$ awk '{print $1","$2;}' data/test.tsv
blah_C1,ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
blah_C2,ACTTTATATATT
blah_C3,ACTTATATATATATA
blah_C4,ACTTATATATATATA
blah_C5,ACTTTATATATT
$ awk '{print $1","$NF;}' data/test.tsv
blah_C1,ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
blah_C2,ACTTTATATATT
blah_C3,ACTTATATATATATA
blah_C4,ACTTATATATATATA
blah_C5,ACTTTATATATT
Awk has two important patterns which are specified by the keyword called BEGIN and END. The syntax is as follows:
BEGIN { Actions before reading the file}
{Actions for everyline in the file}
END { Actions after reading the file }
For example,
$ awk 'BEGIN{print "Header,Sequence"}{print $1","$2;}END{print "-------"}' data/test.tsv
Header,Sequence
blah_C1,ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
blah_C2,ACTTTATATATT
blah_C3,ACTTATATATATATA
blah_C4,ACTTATATATATATA
blah_C5,ACTTTATATATT
-------
We can also use the concept of a conditional operator in print statement of the form print CONDITION ? PRINT_IF_TRUE_TEXT : PRINT_IF_FALSE_TEXT. For example, in the code below, we identify sequences with lengths > 14:
$ awk '{print (length($2)>14) ? $0">14" : $0"<=14";}' data/test.tsv
blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG>14
blah_C2 ACTTTATATATT<=14
blah_C3 ACTTATATATATATA>14
blah_C4 ACTTATATATATATA>14
blah_C5 ACTTTATATATT<=14
We can also use 1 after the last block {} to print everything (1 is a shorthand notation for {print $0} which becomes {print} as without any argument print will print $0 by default), and within this block, we can change $0, for example to assign the first field to $0 for third line (NR==3), we can use:
$ awk 'NR==3{$0=$1}1' data/test.tsv
blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
blah_C2 ACTTTATATATT
blah_C3
blah_C4 ACTTATATATATATA
blah_C5 ACTTTATATATT
You can have as many blocks as you want and they will be executed on each line in the order they appear, for example, if we want to print $1 three times (here we are using printf instead of print as the former doesn't put end-of-line character),
$ awk '{printf $1"\t"}{printf $1"\t"}{print $1}' data/test.tsv
blah_C1 blah_C1 blah_C1
blah_C2 blah_C2 blah_C2
blah_C3 blah_C3 blah_C3
blah_C4 blah_C4 blah_C4
blah_C5 blah_C5 blah_C5
Although, we can also skip executing later blocks for a given line by using next keyword:
$ awk '{printf $1"\t"}NR==3{print "";next}{print $1}' data/test.tsv
blah_C1 blah_C1
blah_C2 blah_C2
blah_C3
blah_C4 blah_C4
blah_C5 blah_C5
$ awk 'NR==3{print "";next}{printf $1"\t"}{print $1}' data/test.tsv
blah_C1 blah_C1
blah_C2 blah_C2
blah_C4 blah_C4
blah_C5 blah_C5
You can also use getline to load the contents of another file in addition to the one you are reading, for example, in the statement given below, the while loop will load each line from test.tsv into k until no more lines are to be read:
$ awk 'BEGIN{while((getline k <"data/test.tsv")>0) print "BEGIN:"k}{print}' data/test.tsv
BEGIN:blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
BEGIN:blah_C2 ACTTTATATATT
BEGIN:blah_C3 ACTTATATATATATA
BEGIN:blah_C4 ACTTATATATATATA
BEGIN:blah_C5 ACTTTATATATT
blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
blah_C2 ACTTTATATATT
blah_C3 ACTTATATATATATA
blah_C4 ACTTATATATATATA
blah_C5 ACTTTATATATT
You can also store data in the memory with the syntax VARIABLE_NAME[KEY]=VALUE which you can later use through for (INDEX in VARIABLE_NAME) command:
$ awk '{i[$1]=1}END{for (j in i) print j"<="i[j]}' data/test.tsv
blah_C1<=1
blah_C2<=1
blah_C3<=1
blah_C4<=1
blah_C5<=1
Given all that you have learned so far, we are going to extract reads from a FASTA file based on IDs supplied in a file. Say, we are given a FASTA file with following contents:
[uzi@quince-srv2 ~/linuxTutorial]$ cat data/test.fa
>blah_C1
ACTGTCTGTC
ACTGTGTTGTG
ATGTTGTGTGTG
>blah_C2
ACTTTATATATT
>blah_C3
ACTTATATATATATA
>blah_C4
ACTTATATATATATA
>blah_C5
ACTTTATATATT
and an IDs file:
[uzi@quince-srv2 ~/linuxTutorial]$ cat data/IDs.txt
blah_C4
blah_C5
After looking at the file, it is immediately clear that the sequences may span multiple lines (for example, for blah_C1). If we want to match an ID, we can first linearize the file by using the conditional operator as discussed above to have the delimited information of each sequence in one line, and then make logic to perform further functionality on each line later. Our logic is that for lines that contain header information /^>/ we can do something differently, and for other lines we use printf to remove new line character:
[uzi@quince-srv2 ~/linuxTutorial]$ awk '{printf /^>/ ? $0 : $0}' data/test.fa
>blah_C1ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG>blah_C2ACTTTATATATT>blah_C3ACTTATATATATATA>blah_C4ACTTATATATATATA>blah_C5ACTTTATATATT
We can then put each sequence on a separate line and also put a tab character ("\t") between the header and the sequence:
[uzi@quince-srv2 ~/linuxTutorial]$ awk '{printf /^>/ ? "\n"$0 : $0}' data/test.fa
>blah_C1ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
>blah_C2ACTTTATATATT
>blah_C3ACTTATATATATATA
>blah_C4ACTTATATATATATA
>blah_C5ACTTTATATATT[uzi@quince-srv2 ~/linuxTutorial]$ awk '{printf /^>/ ? "\n"$0"\t" : $0}' data/test.fa
>blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
>blah_C2 ACTTTATATATT
>blah_C3 ACTTATATATATATA
>blah_C4 ACTTATATATATATA
>blah_C5 ACTTTATATATT
We can then use NR==1 block to stop printing a new line character before the first header (as you can see there is an empty space) and use next to ignore the later block:
[uzi@quince-srv2 ~/linuxTutorial]$ awk 'NR==1{printf $0"\t";next}{printf /^>/ ? "\n"$0"\t" : $0}' data/test.fa
>blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
>blah_C2 ACTTTATATATT
>blah_C3 ACTTATATATATATA
>blah_C4 ACTTATATATATATA
>blah_C5 ACTTTATATATT
We can then pipe this stream to another awk statement using "\t" as a delimeter (which you can specify using -F) and use gsub to remove > from the start of each line since our IDs file doesn't contain that character:
[uzi@quince-srv2 ~/linuxTutorial]$ awk 'NR==1{printf $0"\t";next}{printf /^>/ ? "\n"$0"\t" : $0}' data/test.fa | awk -F"\t" '{gsub("^>","",$0);print $0}'
blah_C1 ACTGTCTGTCACTGTGTTGTGATGTTGTGTGTG
blah_C2 ACTTTATATATT
blah_C3 ACTTATATATATATA
blah_C4 ACTTATATATATATA
blah_C5 ACTTTATATATT
Now we load the IDs.txt file in the BEGIN block, store the IDs in the memory, and in the stream if the first field ($1) matches the ID stored in the memory, we output the formatted record:
[uzi@quince-srv2 ~/linuxTutorial/data]$ awk 'NR==1{printf $0"\t";next}{printf /^>/ ? "\n"$0"\t" : $0}' data/test.fa | awk -F"\t" 'BEGIN{while((getline k < "data/IDs.txt")>0)i[k]=1}{gsub("^>","",$0); if(i[$1]){print ">"$1"\n"$2}}'
>blah_C4
ACTTATATATATATA
>blah_C5
ACTTTATATATT
With Bioawk it is much simpler as you don't have to linearize the FASTA file as the record boundaries are the complete sequence boundaries and not lines:
[uzi@quince-srv2 ~/linuxTutorial/data]$ bioawk -cfastx 'BEGIN{while((getline k <"data/IDs.txt")>0)i[k]=1}{if(i[$name])print ">"$name"\n"$seq}' data/test.fa
>blah_C4
ACTTATATATATATA
>blah_C5
ACTTTATATATT
-c, with the field names as follows (you can use the column pairs alternatively):
bed: $1:$chrom $2:$start $3:$end $4:$name $5:$score $6:$strand $7:$thickstart $8:$thickend $9:$rgb $10:$blockcount $11:$blocksizes $12:$blockstarts sam: $1:$qname $2:$flag $3:$rname $4:$pos $5:$mapq $6:$cigar $7:$rnext $8:$pnext $9:$tlen $10:$seq $11:$qual vcf: $1:$chrom $2:$pos $3:$id $4:$ref $5:$alt $6:$qual $7:$filter $8:$info gff: $1:$seqname $2:$source $3:$feature $4:$start $5:$end $6:$score $7:$filter $8:$strand $9:$group $10:$attribute fastx: $1:$name $2:$seq $3:$qual $4:$comment
Exercise 2: Alignment Statistics for Metagenomics/Population Genomics
For this exercise we will use a C. Difficile Ribotype 078 reference database that comprises of 61 contigs. Even though it is a single genome for which we have obtained the samples, the workflow given below remains similar for the metagenomic samples when you have complete genomes instead of contigs in the reference database (and so I use the nomenclature: genomes/contigs). Before we analyze our samples, we can do some quality control checks on our raw sequences using FastQC. Running the following command will generate a M120_S2_L001_R1_001_fastqc folder with an html page fastqc_report.html inside. You can load it up in your browser to assess your data through graphs and summary tables.
[uzi@quince-srv2 ~/linuxTutorial]$ fastqc data/M120_*R1*.fastq
Started analysis of M120_S2_L001_R1_001.fastq
Approx 5% complete for M120_S2_L001_R1_001.fastq
Approx 10% complete for M120_S2_L001_R1_001.fastq
Approx 15% complete for M120_S2_L001_R1_001.fastq
Approx 20% complete for M120_S2_L001_R1_001.fastq
Approx 25% complete for M120_S2_L001_R1_001.fastq
Approx 30% complete for M120_S2_L001_R1_001.fastq
Approx 35% complete for M120_S2_L001_R1_001.fastq
Approx 40% complete for M120_S2_L001_R1_001.fastq
Approx 45% complete for M120_S2_L001_R1_001.fastq
Approx 50% complete for M120_S2_L001_R1_001.fastq
Approx 55% complete for M120_S2_L001_R1_001.fastq
Approx 60% complete for M120_S2_L001_R1_001.fastq
Approx 65% complete for M120_S2_L001_R1_001.fastq
Approx 70% complete for M120_S2_L001_R1_001.fastq
Approx 75% complete for M120_S2_L001_R1_001.fastq
Approx 80% complete for M120_S2_L001_R1_001.fastq
Approx 85% complete for M120_S2_L001_R1_001.fastq
Approx 90% complete for M120_S2_L001_R1_001.fastq
Approx 95% complete for M120_S2_L001_R1_001.fastq
Approx 100% complete for M120_S2_L001_R1_001.fastq
Analysis complete for M120_S2_L001_R1_001.fastq
[uzi@quince-srv2 ~/linuxTutorial]$
For example, here is the file generated for the above M120_S2_L001_R1_001.fastq file:
Alternatively, you can also try my Shell utilities for QC as well as Shell wrappers for EMBOSS utilities.
Next we index our reference database file. Indexing speeds up alignment, allowing the aligner to quickly find short, near-exact matches to use as seeds for subsequent full-alignments.
[uzi@quince-srv2 ~/linuxTutorial/data]$ bwa index Cdiff078.fa
Use BWA-MEM to align paired-end sequences. Briefly, the algorithm works by seeding alignments with maximal exact matches (MEMs) and then extending seeds with the affine-gap Smith-Waterman algorithm (SW). From BWA doc, it is suggested that for 70bp or longer Illumina, 454, Ion Torrent and Sanger reads, assembly contigs and BAC sequences, BWA-MEM is usually the preferred algorithm. For short sequences, BWA-backtrack may be better. BWA-SW may have better sensitivity when alignment gaps are frequent.
[uzi@quince-srv2 ~/linuxTutorial]$ bwa mem data/Cdiff078.fa data/M120_*R1*.fastq data/M120_*R2*.fastq > aln-pe.sam
We have generated a sam file (aln-pe.sam) which consist of two types of lines: headers and alignments. Headers begin with @, and provide meta-data regarding the entire alignment file. Alignments begin with any character except @, and describe a single alignment of a sequence read against the reference genome. Note that each read in a FASTQ file may align to multiple regions within a reference genome, and an individual read can therefore result in multiple alignments. In the SAM format, each of these alignments is reported on a separate line. Also, each alignment has 11 mandatory fields, followed by a variable number of optional fields. Each of the fields is described in the table below:
| Col | Field | Description |
| 1 | QNAME | Query template/pair NAME |
| 2 | FLAG | bitwise FLAG |
| 3 | RNAME | Reference sequence NAME |
| 4 | POS | 1-based leftmost POSition/coordinate of clipped sequence |
| 5 | MAPQ | MAPping Quality (Phred-scaled) |
| 6 | CIAGR | extended CIGAR string |
| 7 | MRNM | Mate Reference sequence NaMe (‘=’ if same as RNAME) |
| 8 | MPOS | 1-based Mate POSistion |
| 9 | TLEN | inferred Template LENgth (insert size) |
| 10 | SEQ | query SEQuence on the same strand as the reference |
| 11 | QUAL | query QUALity (ASCII-33 gives the Phred base quality) |
| 12+ | OPT | variable OPTional fields in the format TAG:VTYPE:VALUE |
where FLAG is defined as:
| Flag | Chr | Description |
| 0x0001 | p | the read is paired in sequencing |
| 0x0002 | P | the read is mapped in a proper pair |
| 0x0004 | u | the query sequence itself is unmapped |
| 0x0008 | U | the mate is unmapped |
| 0x0010 | r | strand of the query (1 for reverse) |
| 0x0020 | R | strand of the mate |
| 0x0040 | 1 | the read is the first read in a pair |
| 0x0080 | 2 | the read is the second read in a pair |
| 0x0100 | s | the alignment is not primary |
| 0x0200 | f | the read fails platform/vendor quality checks |
| 0x0400 | d | the read is either a PCR or an optical duplicate |
Since the flags are given in decimal representation in the SAM file, you can use this link to check which flag is set. We are going to use SAMTools which provides various tools for manipulating alignments in the SAM/BAM format. The SAM (Sequence Alignment/Map) format (BAM is just the binary form of SAM) is currently the de facto standard for storing large nucleotide sequence alignments. If you are dealing with high-throughput metagenomic whole-genome shotgun sequencing data, you will have to deal with SAM/BAM files. See what SAMtools have to offer:
We can then use a program SAMstat to get statistics on our aln-pe.sam file:
[uzi@quince-srv2 ~/linuxTutorial]$ samstat aln-pe.sam
Running the above code will generate a aln-pe.sam.samstat.html file which you can open in your browser (be patient, it takes a bit of time to load). Plots such as "Reads Length Distributions" and "Base Quality Distributions" may be of interest to you:
Now we convert SAM file to the binary BAM file
[uzi@quince-srv2 ~/linuxTutorial]$ samtools view -h -b -S aln-pe.sam > aln-pe.bam
Extract only those sequences that were mapped against the reference database. Use -F 4 switch.
[uzi@quince-srv2 ~/linuxTutorial]$ samtools view -b -F 4 aln-pe.bam > aln-pe.mapped.bam
Generate a file lengths.genome that contains two entries per row: genome identifier and the corresponding genome length:
[uzi@quince-srv2 ~/linuxTutorial]$ samtools view -H aln-pe.mapped.bam | perl -ne 'if ($_ =~ m/^\@SQ/) { print $_ }' | perl -ne 'if ($_ =~ m/SN:(.+)\s+LN:(\d+)/) { print $1, "\t", $2, "\n"}' > lengths.genome
[uzi@quince-srv2 ~/linuxTutorial]$ cat lengths.genome
Cdiff078_C01 9165
Cdiff078_C02 93786
Cdiff078_C03 752
Cdiff078_C04 5361
Cdiff078_C05 70058
Cdiff078_C06 23538
Cdiff078_C07 98418
Cdiff078_C08 361074
Cdiff078_C09 45183
Cdiff078_C10 141523
Cdiff078_C11 21992
Cdiff078_C12 2353
Cdiff078_C13 133975
Cdiff078_C14 3374
Cdiff078_C15 9744
Cdiff078_C16 25480
Cdiff078_C17 293596
Cdiff078_C18 7057
Cdiff078_C19 73989
Cdiff078_C20 248092
Cdiff078_C21 41937
Cdiff078_C22 65693
Cdiff078_C23 21321
Cdiff078_C24 440055
Cdiff078_C25 210910
Cdiff078_C26 164162
Cdiff078_C27 22782
Cdiff078_C28 201701
Cdiff078_C29 13447
Cdiff078_C30 101704
Cdiff078_C31 146436
Cdiff078_C32 61153
Cdiff078_C33 59640
Cdiff078_C34 193273
Cdiff078_C35 18395
Cdiff078_C36 25573
Cdiff078_C37 61616
Cdiff078_C38 4117
Cdiff078_C39 110461
Cdiff078_C40 125351
Cdiff078_C41 38508
Cdiff078_C42 113221
Cdiff078_C43 500
Cdiff078_C44 547
Cdiff078_C45 613
Cdiff078_C46 649
Cdiff078_C47 666
Cdiff078_C48 783
Cdiff078_C49 872
Cdiff078_C50 872
Cdiff078_C51 879
Cdiff078_C52 921
Cdiff078_C53 955
Cdiff078_C54 1217
Cdiff078_C55 1337
Cdiff078_C56 1445
Cdiff078_C57 2081
Cdiff078_C58 2098
Cdiff078_C59 2512
Cdiff078_C60 2800
Cdiff078_C61 4372
[uzi@quince-srv2 ~/linuxTutorial]$
Sort BAM file. Many of the downstream analysis programs that use BAM files actually require a sorted BAM file. -m specifies the maximum memory to use, and can be changed to fit your system.
[uzi@quince-srv2 ~/linuxTutorial]$ samtools sort -m 1000000000 aln-pe.mapped.bam aln-pe.mapped.sorted
We will now use bedtools. It is a very useful suite of programs for working with SAM/BAM, BED, VCF and GFF files, files that you will encouter many times doing NGS analysis. -ibam switch takes indexed bam file that we generated earlier, -d reports the depth at each genome position with 1-based coordinates, and -g is used to provide the genome lengths file we generated earlier. The coverage flags are explained pictorially from genomecov man page: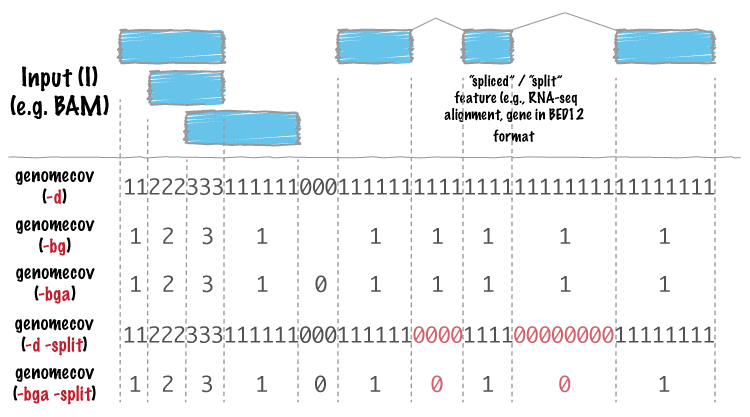
Reference: http://bedtools.readthedocs.org/en/latest/_images/genomecov-glyph.png
[uzi@quince-srv2 ~/linuxTutorial]$ bedtools genomecov -ibam aln-pe.mapped.sorted.bam -d -g lengths.genome > aln-pe.mapped.bam.perbase.cov
Look at the first few entries in the file generated above. First column is genome identifier, second column is position on genome, and third column is coverage.
[uzi@quince-srv2 ~/linuxTutorial]$ head aln-pe.mapped.bam.perbase.cov
Cdiff078_C01 1 41
Cdiff078_C01 2 41
Cdiff078_C01 3 42
Cdiff078_C01 4 42
Cdiff078_C01 5 42
Cdiff078_C01 6 44
Cdiff078_C01 7 44
Cdiff078_C01 8 44
Cdiff078_C01 9 44
Cdiff078_C01 10 44
[uzi@quince-srv2 ~/linuxTutorial]$
Now we will count only those positions where we have >0 coverage.
[uzi@quince-srv2 ~/linuxTutorial]$ awk -F"\t" '$3>0{print $1}' aln-pe.mapped.bam.perbase.cov | sort | uniq -c > aln-pe.mapped.bam.perbase.count
To see what we have done, use the cat command
[uzi@quince-srv2 ~/linuxTutorial]$ cat aln-pe.mapped.bam.perbase.count
9165 Cdiff078_C01
93786 Cdiff078_C02
752 Cdiff078_C03
5361 Cdiff078_C04
70058 Cdiff078_C05
23538 Cdiff078_C06
98418 Cdiff078_C07
333224 Cdiff078_C08
44803 Cdiff078_C09
141523 Cdiff078_C10
21969 Cdiff078_C11
2292 Cdiff078_C12
133974 Cdiff078_C13
1762 Cdiff078_C14
50 Cdiff078_C15
10232 Cdiff078_C16
293440 Cdiff078_C17
7057 Cdiff078_C18
73989 Cdiff078_C19
248092 Cdiff078_C20
41937 Cdiff078_C21
65447 Cdiff078_C22
21321 Cdiff078_C23
439123 Cdiff078_C24
210910 Cdiff078_C25
164162 Cdiff078_C26
22782 Cdiff078_C27
201701 Cdiff078_C28
13447 Cdiff078_C29
98510 Cdiff078_C30
146261 Cdiff078_C31
61153 Cdiff078_C32
44523 Cdiff078_C33
193180 Cdiff078_C34
18395 Cdiff078_C35
25573 Cdiff078_C36
61616 Cdiff078_C37
4117 Cdiff078_C38
62897 Cdiff078_C39
125351 Cdiff078_C40
38508 Cdiff078_C41
113221 Cdiff078_C42
442 Cdiff078_C43
649 Cdiff078_C46
663 Cdiff078_C47
766 Cdiff078_C48
580 Cdiff078_C51
1110 Cdiff078_C54
1445 Cdiff078_C56
2512 Cdiff078_C59
2800 Cdiff078_C60
[uzi@quince-srv2 ~/linuxTutorial]$
We will now use the above file with lengths.genome to calculate the proportions of genomes/contigs covered using the following one-liner. It reads lengths.genome line by line, assigns the genome identifier to myArray[0], it's length tomyArray[1]. It then searches the identifier in aln-pe.mapped.bam.perbase.count, extracts the base count, and uses bc to calculate the proportions.
[uzi@quince-srv2 ~/linuxTutorial]$ while IFS=$'\t' read -r -a myArray; do echo -e "${myArray[0]},$( echo "scale=5;0"$(awk -v pattern="${myArray[0]}" '$2==pattern{print $1}' aln-pe.mapped.bam.perbase.count)"/"${myArray[1]} | bc ) "; done < lengths.genome > aln-pe.mapped.bam.genomeproportion
[uzi@quince-srv2 ~/linuxTutorial]$ cat aln-pe.mapped.bam.genomeproportion
Cdiff078_C01,1.00000
Cdiff078_C02,1.00000
Cdiff078_C03,1.00000
Cdiff078_C04,1.00000
Cdiff078_C05,1.00000
Cdiff078_C06,1.00000
Cdiff078_C07,1.00000
Cdiff078_C08,.92286
Cdiff078_C09,.99158
Cdiff078_C10,1.00000
Cdiff078_C11,.99895
Cdiff078_C12,.97407
Cdiff078_C13,.99999
Cdiff078_C14,.52222
Cdiff078_C15,.00513
Cdiff078_C16,.40156
Cdiff078_C17,.99946
Cdiff078_C18,1.00000
Cdiff078_C19,1.00000
Cdiff078_C20,1.00000
Cdiff078_C21,1.00000
Cdiff078_C22,.99625
Cdiff078_C23,1.00000
Cdiff078_C24,.99788
Cdiff078_C25,1.00000
Cdiff078_C26,1.00000
Cdiff078_C27,1.00000
Cdiff078_C28,1.00000
Cdiff078_C29,1.00000
Cdiff078_C30,.96859
Cdiff078_C31,.99880
Cdiff078_C32,1.00000
Cdiff078_C33,.74652
Cdiff078_C34,.99951
Cdiff078_C35,1.00000
Cdiff078_C36,1.00000
Cdiff078_C37,1.00000
Cdiff078_C38,1.00000
Cdiff078_C39,.56940
Cdiff078_C40,1.00000
Cdiff078_C41,1.00000
Cdiff078_C42,1.00000
Cdiff078_C43,.88400
Cdiff078_C44,0
Cdiff078_C45,0
Cdiff078_C46,1.00000
Cdiff078_C47,.99549
Cdiff078_C48,.97828
Cdiff078_C49,0
Cdiff078_C50,0
Cdiff078_C51,.65984
Cdiff078_C52,0
Cdiff078_C53,0
Cdiff078_C54,.91207
Cdiff078_C55,0
Cdiff078_C56,1.00000
Cdiff078_C57,0
Cdiff078_C58,0
Cdiff078_C59,1.00000
Cdiff078_C60,1.00000
Cdiff078_C61,0
We have a total of 61 genomes/contigs in the reference database. To see how many genomes/contigs we recovered, we will use the following one-liner:
[uzi@quince-srv2 ~/linuxTutorial]$ awk -F "," '{sum+=$NF} END{print "Total genomes covered:"sum}' aln-pe.mapped.bam.genomeproportion
Total genomes covered:47.5224
We also need genome/contig coverage, which we can calculate as:
[uzi@quince-srv2 ~/linuxTutorial]$ bedtools genomecov -ibam aln-pe.mapped.sorted.bam -g lengths.genome | awk -F"\t" '!/^genome/{l[$1]=l[$1]+($2 *$3);r[$1]=$4} END {for (i in l){print i","(l[i]/r[i])}}' > aln-pe.mapped.bam.genomecoverage
[uzi@quince-srv2 ~/linuxTutorial]$ cat aln-pe.mapped.bam.genomecoverage
Cdiff078_C10,61.5467
Cdiff078_C11,68.9158
Cdiff078_C12,79.7875
Cdiff078_C13,61.2645
Cdiff078_C14,57.3438
Cdiff078_C15,0.0812808
Cdiff078_C16,23.5227
Cdiff078_C17,57.358
Cdiff078_C30,59.3333
Cdiff078_C18,55.5597
Cdiff078_C31,62.147
Cdiff078_C19,56.3139
Cdiff078_C32,66.0493
Cdiff078_C33,48.8165
Cdiff078_C34,65.7106
Cdiff078_C35,62.7728
Cdiff078_C36,62.7535
Cdiff078_C37,67.2169
Cdiff078_C51,1.05916
Cdiff078_C38,61.9871
Cdiff078_C39,37.3289
Cdiff078_C54,6.46754
Cdiff078_C56,815.224
Cdiff078_C59,801.998
Cdiff078_C01,67.3333
Cdiff078_C02,67.4621
Cdiff078_C03,103.848
Cdiff078_C04,65.4128
Cdiff078_C05,66.1244
Cdiff078_C06,66.239
Cdiff078_C07,76.0081
Cdiff078_C20,55.6661
Cdiff078_C08,60.4236
Cdiff078_C21,56.2321
Cdiff078_C09,76.9986
Cdiff078_C22,56.8815
Cdiff078_C23,53.2772
Cdiff078_C24,56.9991
Cdiff078_C25,57.4446
Cdiff078_C26,59.296
Cdiff078_C40,66.0074
Cdiff078_C27,59.4391
Cdiff078_C41,67.5941
Cdiff078_C28,59.8319
Cdiff078_C42,69.4415
Cdiff078_C29,60.961
Cdiff078_C43,4.812
Cdiff078_C46,29.3837
Cdiff078_C60,62.1336
Cdiff078_C47,7.95946
Cdiff078_C48,15.3436
[uzi@quince-srv2 ~/linuxTutorial]$
Sort the original bam file
[uzi@quince-srv2 ~/linuxTutorial]$ samtools sort -m 1000000000 aln-pe.bam aln-pe.sorted
Now we will check alignment statistics using the Picard tools. Note that the awk statement given below is used to transpose the original table and you can do without it.
[uzi@quince-srv2 ~/linuxTutorial]$ java -jar $(which CollectAlignmentSummaryMetrics.jar) INPUT=aln-pe.sorted.bam OUTPUT=aln-pe.sorted.alignment_stats.txt REFERENCE_SEQUENCE=data/Cdiff078.fa
[uzi@quince-srv2 ~/linuxTutorial]$ grep -vi -e "^#" -e "^$" aln-pe.sorted.alignment_stats.txt | awk -F"\t" '{ for (i=1; i<=NF; i++) {a[NR,i] = $i}}NF>p{p=NF}END{for(j=1;j<=p;j++){str=a[1,j];for(i=2; i<=NR; i++){str=str"\t"a[i,j];} print str}}'
CATEGORY FIRST_OF_PAIR SECOND_OF_PAIR PAIR
TOTAL_READS 425271 425038 850309
PF_READS 425271 425038 850309
PCT_PF_READS 1 1 1
PF_NOISE_READS 0 0 0
PF_READS_ALIGNED 407011 405258 812269
PCT_PF_READS_ALIGNED 0.957063 0.953463 0.955263
PF_ALIGNED_BASES 119451610 118113100 237564710
PF_HQ_ALIGNED_READS 401018 399295 800313
PF_HQ_ALIGNED_BASES 118606615 117274833 235881448
PF_HQ_ALIGNED_Q20_BASES 116971078 111640501 228611579
PF_HQ_MEDIAN_MISMATCHES 0 0 0
PF_MISMATCH_RATE 0.002359 0.007186 0.004759
PF_HQ_ERROR_RATE 0.002269 0.007065 0.004653
PF_INDEL_RATE 0.000124 0.00013 0.000127
MEAN_READ_LENGTH 299.093366 298.832657 298.963048
READS_ALIGNED_IN_PAIRS 404714 404545 809259
PCT_READS_ALIGNED_IN_PAIRS 0.994356 0.998241 0.996294
BAD_CYCLES 0 0 0
STRAND_BALANCE 0.500072 0.500484 0.500278
PCT_CHIMERAS 0.014823 0.014668 0.014746
PCT_ADAPTER 0.000285 0.000261 0.000273
SAMPLE
LIBRARY
READ_GROUP
[uzi@quince-srv2 ~/linuxTutorial]$
The detailed description of these summary metrics are given here. From this link, PF_MISMATCH_RATE, PF_HQ_ERROR_RATE, and PF_INDEL_RATE are of interest to us. As can be seen, the error rates are quite low and we can proceed with the analysis. Next we would like to calculate GC bias. For this purpose, we will index aln-pe.mapped.sorted.bam file.
[uzi@quince-srv2 ~/linuxTutorial]$ samtools index aln-pe.mapped.sorted.bam
[uzi@quince-srv2 ~/linuxTutorial]$ for i in $(samtools view -H aln-pe.mapped.sorted.bam | awk -F"\t" '/@SQ/{gsub("^SN:","",$2);print $2}'
);do samtools view -b aln-pe.mapped.sorted.bam $i > aln-pe.mapped.sorted.$i.bam; java -Xmx2g -jar $(which CollectGcBiasMetrics.jar) R=data/Cdiff078.fa I=aln-pe.mapped.sorted.$i.bam O=aln-pe.mapped.sorted.${i}_GCBias.txt CHART=aln-pe.mapped.sorted.${i}_GCBias.pdf ASSUME_SORTED=true; done
In the above one-liner, CollectGcBiasMetrics.jar will generate a GC bias plot for each contig, and will look like these: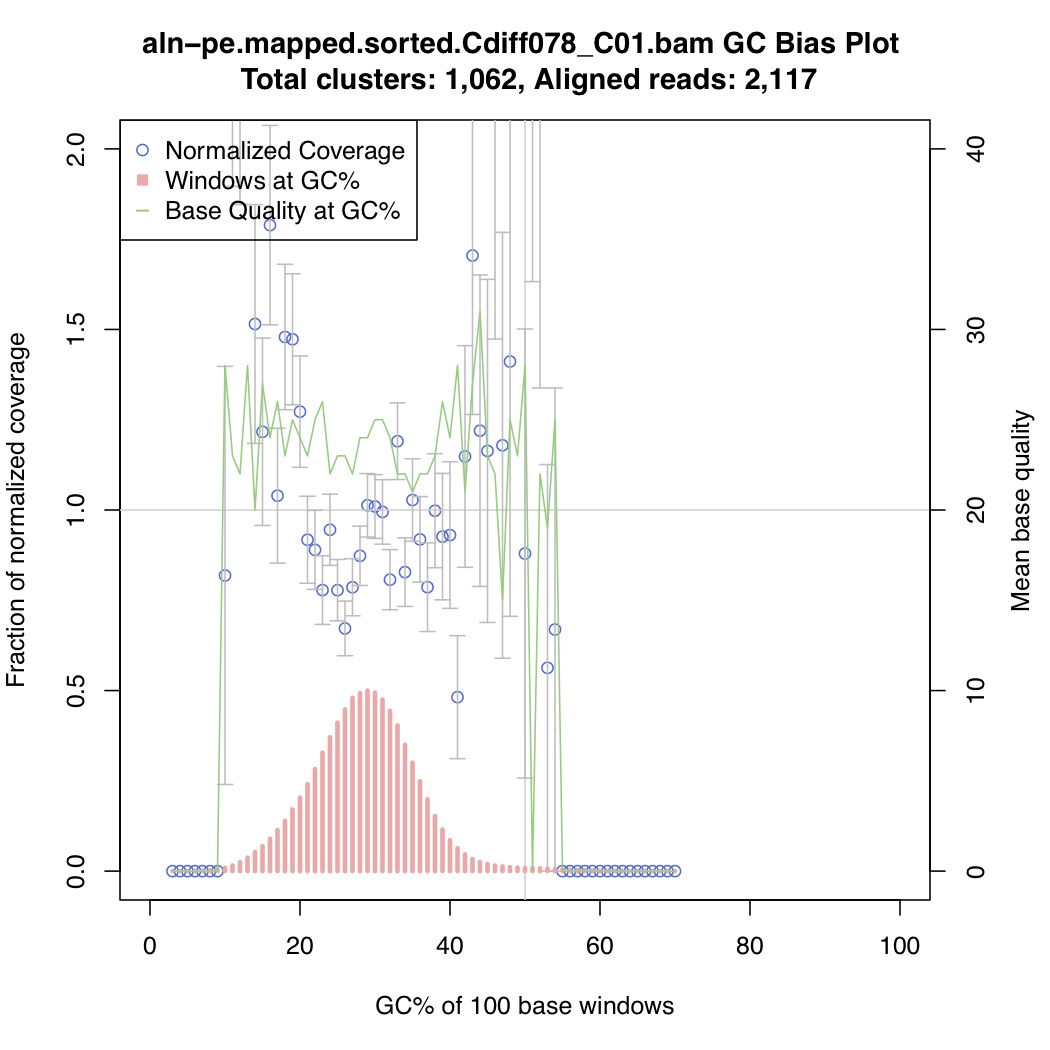
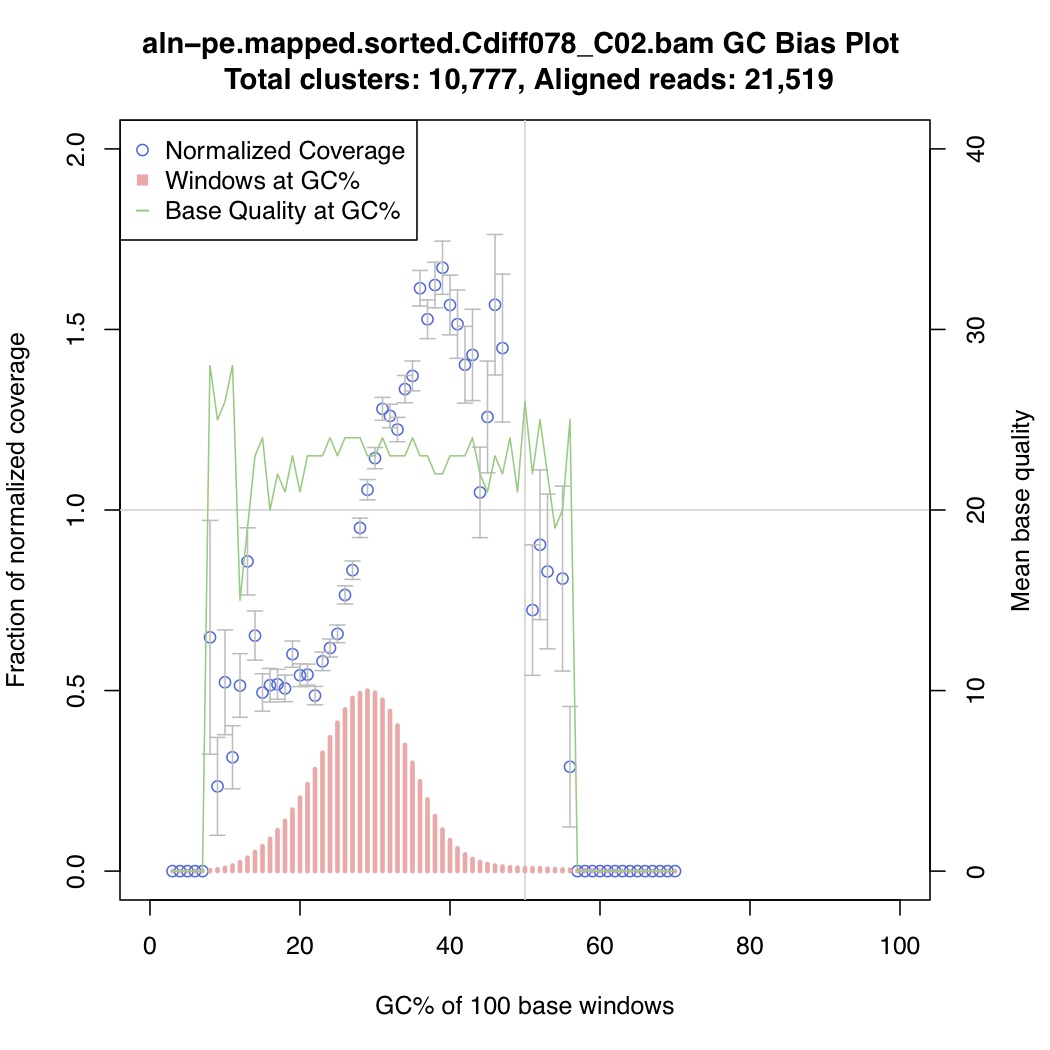
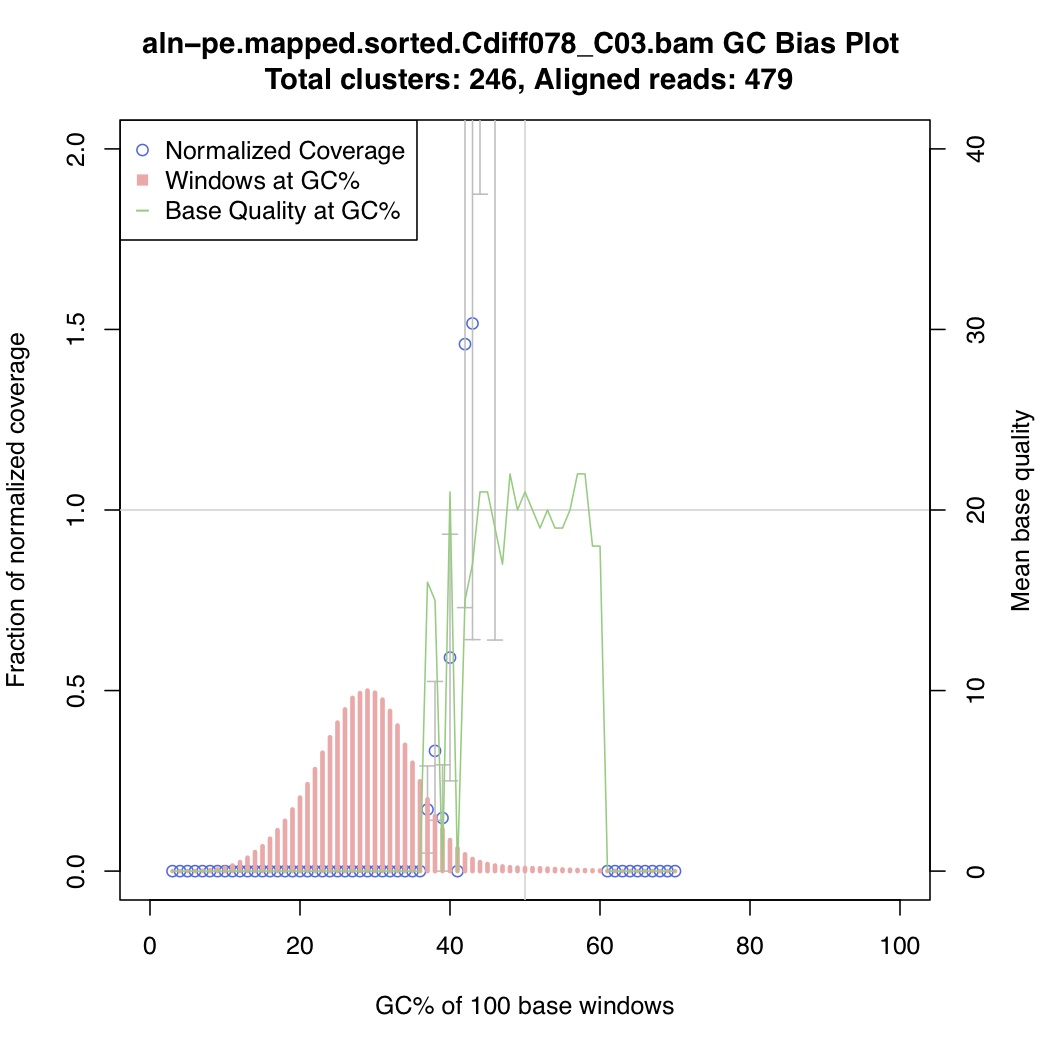
Now collate all the txt files together:
[uzi@quince-srv2 ~/linuxTutorial]$ for i in $(ls *_GCBias.txt); do awk -v k="$i" '!/^#/ && !/^$/ && !/^GC/ && !/?/{print k"\t"$1"\t"$5}' $i; done | perl -ane '$r{$F[0].":".$F[1]}=$F[2];unless($F[0]~~@s){push @s,$F[0];}unless($F[1]~~@m){push @m,$F[1];}END{print "Contigs\t".join("\t",@s)."\n";for($i=0;$i<@m;$i++){print $m[$i];for($j=0;$j<@s;$j++){(not defined $r{$s[$j].":".$m[$i]})?print "\t".0:print"\t".$r{$s[$j].":".$m[$i]};}print "\n";}}' | sed '1s/aln-pe\.mapped\.sorted\.//g;1s/_GCBias\.txt//g' > aln-pe.mapped.sorted.bam.gcbias
GENERATEtable.sh script. If the data stream is of the form [Contig]\t[Feature]\t[Value], then you can pipe the stream to GENERATEtable.sh to obtain a Contig X Feature table:
$ cat test.tsv
contig1 F1 12.2
contig1 F2 34.2
contig1 F3 45.2
contig2 F2 56.3
contig2 F3 56.2
contig3 F1 45.4
contig3 F2 56.3
contig4 F1 23.5
contig5 F1 24.5
$ cat GENERATEtable.sh
#!/bin/bash
less <&0| \
perl -ane '$r{$F[0].":".$F[1]}=$F[2];
unless($F[0]~~@s){
push @s,$F[0];}
unless($F[1]~~@m){
push @m,$F[1];}
END{
print "Contigs\t".join("\t",@s)."\n";
for($i=0;$i<@m;$i++){
print $m[$i];
for($j=0;$j<@s;$j++){
(not defined $r{$s[$j].":".$m[$i]})?print "\t".0:print"\t".$r{$s[$j].":".$m[$i]};}
print "\n";}}'
$ cat test.tsv | ./GENERATEtable.sh
Contigs contig1 contig2 contig3 contig4 contig5
F1 12.2 0 45.4 23.5 24.5
F2 34.2 56.3 56.3 0 0
F3 45.2 56.2 0 0 0
Now take a look at the generated table:
[uzi@quince-srv2 ~/linuxTutorial]$ head aln-pe.mapped.sorted.bam.gcbias
Contigs Cdiff078_C01 Cdiff078_C02 Cdiff078_C03 Cdiff078_C04 Cdiff078_C05 Cdiff078_C06 Cdiff078_C07 Cdiff078_C08 Cdiff078_C09 Cdiff078_C10 Cdiff078_C11 Cdiff078_C12 Cdiff078_C13 Cdiff078_C14 Cdiff078_C15 Cdiff078_C16 Cdiff078_C17 Cdiff078_C18 Cdiff078_C19 Cdiff078_C20 Cdiff078_C21 Cdiff078_C22 Cdiff078_C23 Cdiff078_C24 Cdiff078_C25 Cdiff078_C26 Cdiff078_C27 Cdiff078_C28 Cdiff078_C29 Cdiff078_C30 Cdiff078_C31 Cdiff078_C32 Cdiff078_C33 Cdiff078_C34 Cdiff078_C35 Cdiff078_C36 Cdiff078_C37 Cdiff078_C38 Cdiff078_C39 Cdiff078_C40 Cdiff078_C41 Cdiff078_C42 Cdiff078_C43 Cdiff078_C46 Cdiff078_C47 Cdiff078_C48 Cdiff078_C51 Cdiff078_C54 Cdiff078_C56 Cdiff078_C59 Cdiff078_C60
3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
4 0 0 0 0 0 0 0 0 0 0 0 0 19.855622 0 113.514035 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
5 0 0 0 0 0 0 0 0 0 5.94012 0 0 9.265957 0 21.189287 0 0 1.013634 0 0 0 0 0 0 0 0 0 0 21.79477 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
6 0 0 0 0 0 0 0 0 0 2.099257 0 0 8.004623 17.472841 0 0 4.059842 0.50366 0 0 1.011679 0 0 3.954422 0 15.404681 0 4.36693 0 1.355168 0 0 0 0 0 0 0 0 0 0
7 0 0 0 0 1.898603 0 0 0 5.621592 3.322151 0 0 1.063016 0 0 0 0.395449 0 0 1.454454 11.850605 1.167889 0 2.616454 0.919685 0.229238 0 0.554199 0 0 2.647623 1.10446 0 0.699548 5.625801 0 3.721214 0 1.484725 0 0 0 0 0 0 0 0 0 23.930557
8 0 0.647487 0 0 5.277073 0 1.787893 0 4.918962 1.302196 5.292116 0 0.73865 2.481033 0 0 0.488503 0 0 1.796704 8.023401 0.270507 0 2.181692 1.27811 1.168119 0 1.625944 0 0 1.672486 1.534896 0.32406 4.34351 0 0.738781 0 0.917049 0 0 0.646285 0 0 0 0 0
9 0 0.2347 0 0 1.275217 0 0.465283 0.362786 3.35627 0.457715 2.237992 0 1.725467 4.796381 0 0 0.885359 0 0.719303 1.9538 3.673656 0.915163 1.311588 1.97216 0.494173 1.283086 0 0.909904 0 0.080909 1.131649 2.225468 0 0.352394 1.259544 0 1.071169 13.719311 1.329643 0.540768 0.765043 0.562234 0 0 0 0 0 0 0 0 8.036611
10 0.818789 0.522956 0 0 1.092856 0.491785 0.512673 0.769493 1.150524 0.794326 2.959123 0 1.437926 1.849716 0 0 0.713225 0.63983 0.739728 1.748817 2.30876 1.411724 0.674416 1.696827 1.355215 1.504252 0.187891 1.361078 0 0.582441 0.997528 2.288661 0 1.147602 0 1.42304 1.223984 4.409023 0.569749 0.463436 0.393384 0.321222 0 0 0 0 0 0 0 0
11 2.714431 0.315217 0 0 0.724603 0.494047 0.247215 0.885474 1.083578 0.904377 2.378187 0 1.954739 0 0 0.50476 0.713456 0 1.226165 1.513891 1.138604 2.795913 1.355035 1.755064 1.697555 1.208937 0.566266 1.204967 0 0.927834 1.018818 1.456152 0 1.04366 0.260253 0.857751 0.848433 0.892897 0.335209 0.829905 0.755116 0 0 0 0 0 0 0 0 2.490848
You can then use the following R code to generate a GC vs Coverage table which shows that at very GC, coverages go down (note that these are the smoothed values across all genomes/contigs):
library(ggplot2) library(reshape) data_table <- read.csv("aln-pe.mapped.sorted.bam.gcbias",header=TRUE,row.names=1,sep="\t") df<-NULL for(i in names(data_table)){ tmp<-data.frame(rownames(data_table),data_table[,i],rep(i,dim(data_table)[1])) if(is.null(df)){df<-tmp}else{df<-rbind(df,tmp)} } names(df)<-c("GC","Coverage","Contigs") df$GC<-as.numeric(df$GC) p<-ggplot(df,aes(GC,Coverage,group=Contigs)) + geom_smooth(aes(group="dummy"),method = "loess", formula = y ~ x, size = 1)+ theme_bw()+ theme(axis.text.x=element_text(angle=90,hjust=1,vjust=0.5))+ pdf("aln-pe.mapped.sorted.bam.gcbias.pdf") print(p) dev.off()
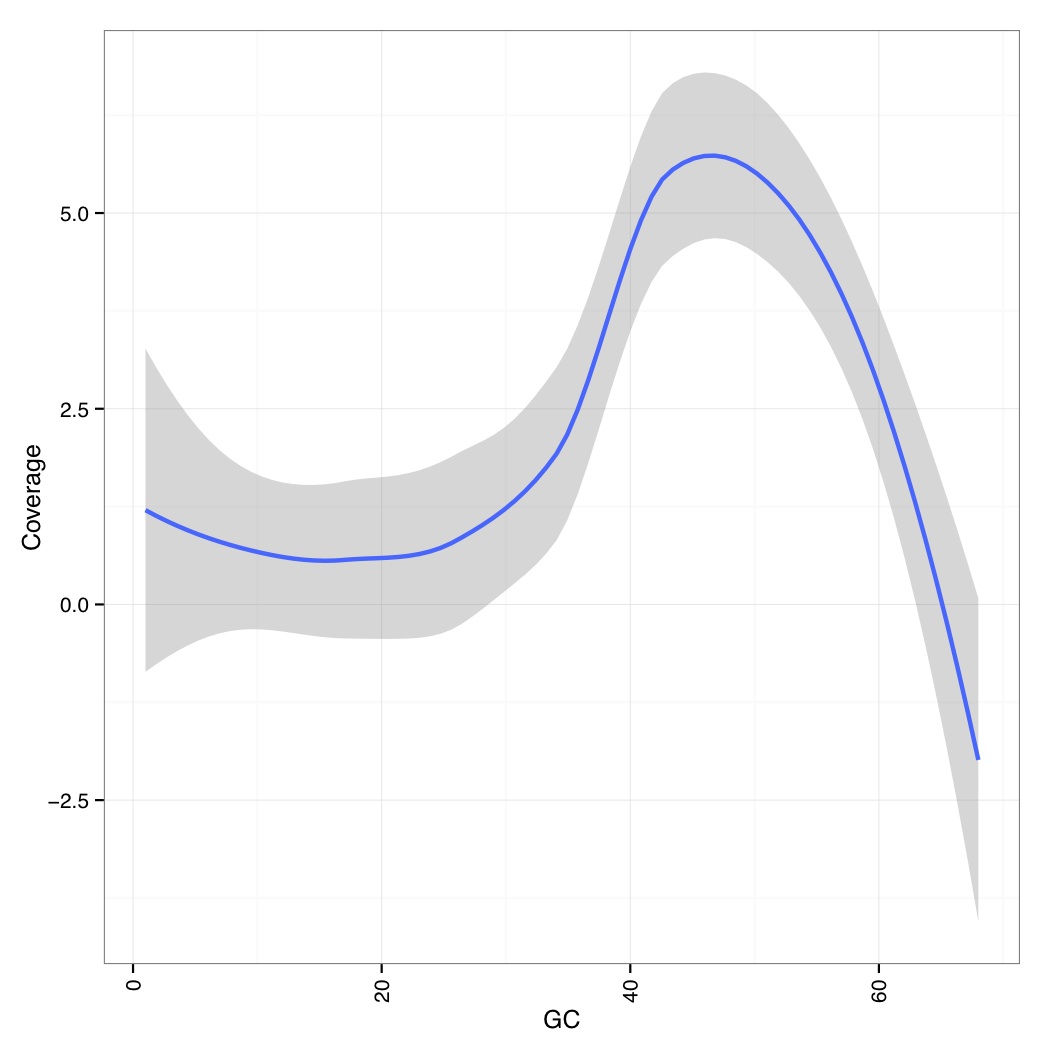
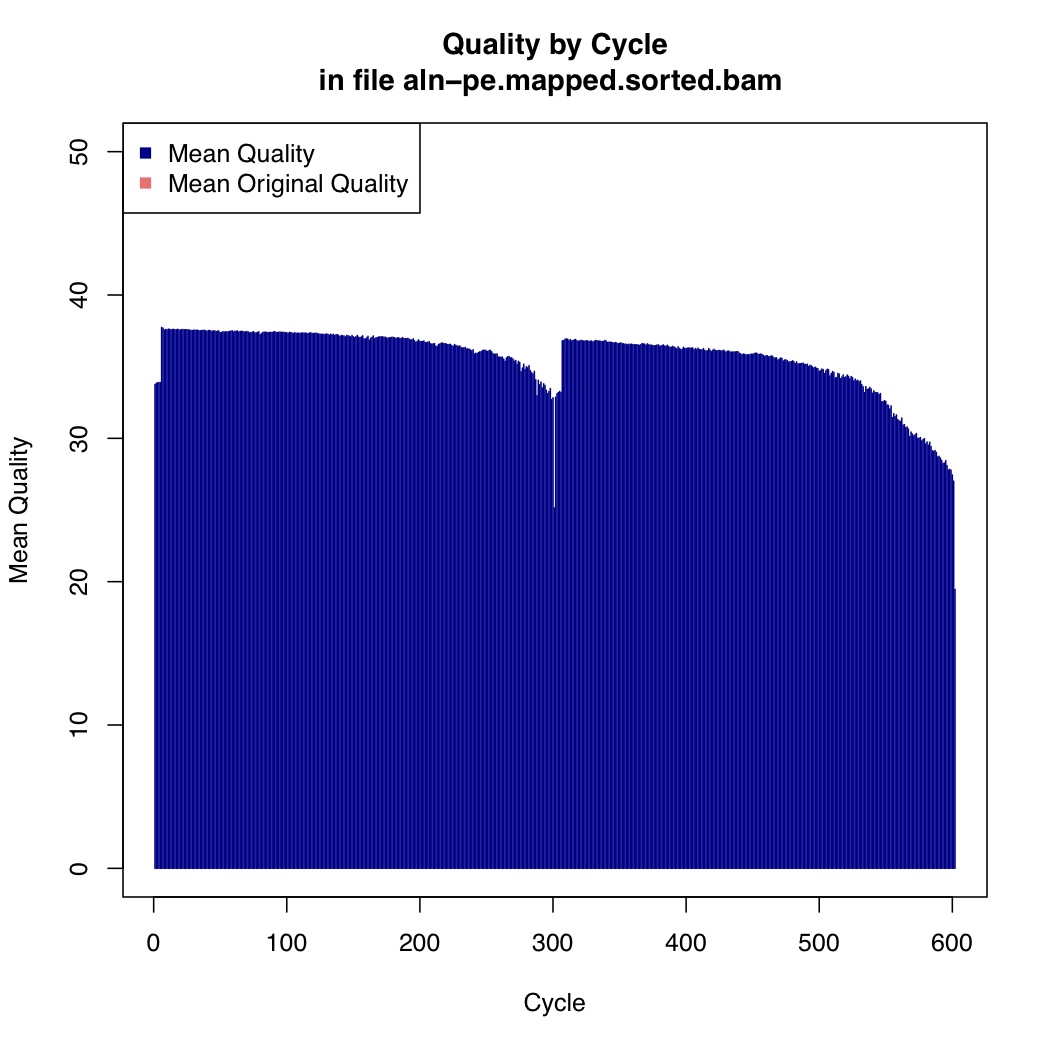
Now we calculate mean quality score by cycle
[uzi@quince-srv2 ~/linuxTutorial]$ java -Xmx2g -jar $(which MeanQualityByCycle.jar) INPUT=aln-pe.mapped.sorted.bam OUTPUT=aln-pe.mapped.sorted.mqc.txt CHART_OUTPUT=aln-pe.mapped.sorted.mqc.pdf
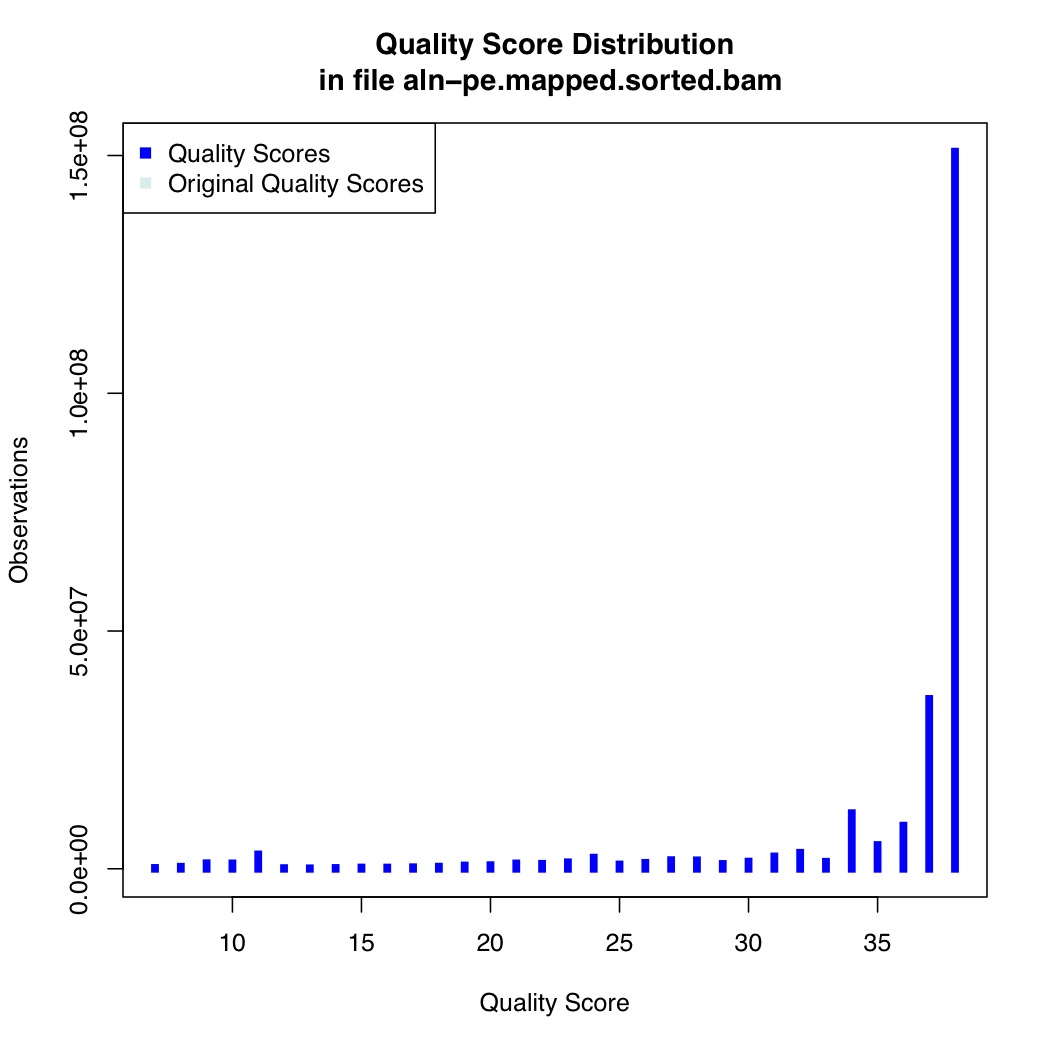
We also calculate quality score distribution
[uzi@quince-srv2 ~/linuxTutorial]$ java -Xmx2g -jar $(which QualityScoreDistribution.jar) INPUT=aln-pe.mapped.sorted.bam OUTPUT=aln-pe.mapped.sorted.qsd.txt CHART_OUTPUT=aln-pe.mapped.sorted.qsd.pdf
Another useful tool is Qualimap which offers Multi-sample BAM QC.
To use it, we need to generate input.txt file which contains listing of BAM files we want to compare. To save time, I am only considering 3 files:
[uzi@quince-srv2 ~/linuxTutorial]$ awk '{split($0,k,".");print k[4]"\t"$0}' <(ls *.sorted.Cdiff078_C4*.bam | head -3) > input.txt
[uzi@quince-srv2 ~/linuxTutorial]$ cat input.txt
Cdiff078_C40 aln-pe.mapped.sorted.Cdiff078_C40.bam
Cdiff078_C41 aln-pe.mapped.sorted.Cdiff078_C41.bam
Cdiff078_C42 aln-pe.mapped.sorted.Cdiff078_C42.bam
[uzi@quince-srv2 ~/linuxTutorial]$ qualimap multi-bamqc -d input.txt -r -outdir qualimap
This will generate a qualimap folder that will contain a multisampleBamQcReport.html file that you can load in your browser. Do check the "PCA" and "Insert Size Histogram" plot.
You can use samtools flagstat to get mapping statistics:
[uzi@quince-srv2 ~/linuxTutorial]$ samtools flagstat aln-pe.bam
850309 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 duplicates
812269 + 0 mapped (95.53%:-nan%)
850309 + 0 paired in sequencing
425271 + 0 read1
425038 + 0 read2
795935 + 0 properly paired (93.61%:-nan%)
809259 + 0 with itself and mate mapped
3010 + 0 singletons (0.35%:-nan%)
11922 + 0 with mate mapped to a different chr
8256 + 0 with mate mapped to a different chr (mapQ>=5)
With Bioawk, you can do amazing things:
Extracting unmapped reads without headers:
[uzi@quince-srv2 ~/linuxTutorial]$ bioawk -c sam 'and($flag,4)' aln-pe.sam | less
M01808:26:000000000-A6KTK:1:1101:10450:1106 77 * 0 0 * * 0 0 TTAAAGTTAAACTTGTCATATTCATATCTGATTTTTCTACTAGATTCCTTTAAGTTATCCGAACATGAAGCAAGTAATTTATCCTTAATTAAATTATAGACTTTACTTTCTTTATCAGATAAATCTTTAGCTTTTCCAATACCAGATATAGTAGGAATAATTGCATAGTGGTCTGTAACTTTAGATGAATTAAAAATAGACTTAAAGTTTGATTCATTGATTTTAAAATCTTCTTCAAGTCCTTCTAATAATTCTTTCATAGTATTAACCATATCATTGGTTAAATACCTGCTATCCGTTC CCCCCGGGGGGGGGGFGFGGGGGGGGGGGGGCEGGGGGFGGGGEFEEGGFEGGGDFFGFGGGFFFGGGGGGFFFGGGGFEFF<fee@cffgc@ffcefgfdfcggg<cfdfgcfgfgadgefafggfggfggggggg<efdgfa<fgffgfffdcefg9egfgffgcffeafgg@fdfggggggcfe=efgggggffffggge9@=egggffffefgggddfgffgg,edfgggggfggggfgggggdfgggggggdgggdfg7ffcffcgf?cc7cffffffaccefafgfa@e5@e8@) as:i:0="" xs:i:0="" m01808:26:000000000-a6ktk:1:1101:10450:1106="" 141="" *="" 0="" gatctcactaccttacaaagagagtcaaataaatattttgggtattcagcaaatgatactttaaatctagcacaaagtttgtatgaaaagaagctaatcacatatccaagcacggatagcaggtatttaaccactgatatggttaatactatgaaagaattattagaaggacttgaagaagattttaaaatcaatgaatcaaactttaagtctatttttaattcatctaaagttacagaccactatgcaattattcctactctatctggtattggaaaagctaaagatttatctgataa="" cccccgggfffggggfgg@dggggggggfgggggggfggggggggffcefgfffgcefgggggggggga@fgggd8ffgggggggfggg8ffgf8egggggggfgggggf,b@bc@cceggggggggggggff,efggdfgggg9ffgggggggggd,9efgggggf9="">EBFGGGC>FGGFBFGGGFDF,@DEEFCFGGGGGGGGCEF,?EGFGGGFDFGGFGGFDCDFFGFFDFFFBFBFFGFFGEFFAAFCEFFFFF5@09*2?EE@A*>@AEF5@):=>E;EB**9:495* AS:i:0 XS:i:0
M01808:26:000000000-A6KTK:1:1101:10136:1113 77 * 0 0 * * 0 0 CTATTGGAACAAGTGGGGAACTGCAGTCGCCTAACAGAGAATATATTCGTTATCGAATTACATTATCTACTCAAGACACCAGTAGAACTCCTAAACTTCTTGAAATACAACTACATGATATACCAAAACCTCCTTATGAGAGACTTGGATTTGCAAGACCAGTTGTGTTGGATACTAACGGGGCTTGGGAAGCAGTGTTAGAAAATGCCTTTGATATTGTAGTAACAAGTGAAGTAAATGGCGCTGATATTCTGGAGTTTAAACTGCCATTTCATGATTCCAAGCGAGAGACATTAGACA CCCCCGGGGEGGF<fffeggffggggfegfgcggggafgffgggfgfcfgggggfgggggggggggggggggggggggggfgggggggggggfggg:< span="">
Extracting mapped reads with headers:
[uzi@quince-srv2 ~/linuxTutorial]$ bioawk -c sam -H '!and($flag,4)' aln-pe.sam | less
@SQ SN:Cdiff078_C01 LN:9165
@SQ SN:Cdiff078_C02 LN:93786
@SQ SN:Cdiff078_C03 LN:752
@SQ SN:Cdiff078_C04 LN:5361
@SQ SN:Cdiff078_C05 LN:70058
@SQ SN:Cdiff078_C06 LN:23538
@SQ SN:Cdiff078_C07 LN:98418
@SQ SN:Cdiff078_C08 LN:361074
@SQ SN:Cdiff078_C09 LN:45183
@SQ SN:Cdiff078_C10 LN:141523
@SQ SN:Cdiff078_C11 LN:21992
@SQ SN:Cdiff078_C12 LN:2353
@SQ SN:Cdiff078_C13 LN:133975
@SQ SN:Cdiff078_C14 LN:3374
@SQ SN:Cdiff078_C15 LN:9744
@SQ SN:Cdiff078_C16 LN:25480
:
Create FASTA from BAM (uses revcomp if FLAG & 16):
[uzi@quince-srv2 ~/linuxTutorial]$ samtools view aln-pe.bam | bioawk -c sam '{ s=$seq; if(and($flag, 16)) {s=revcomp($seq) } print ">"$qname"\n"s}' | less
>M01808:26:000000000-A6KTK:1:1101:19201:1002
NAAAAGAACTGGCAATTGAAAATAATATACCTGTATATCAACCAGTAAAGGCTAGAGATAAAGAATTTATAGATACAATTAAATCTTTAAATCCAGATGTAATAGTAGTTGTAGCTTTTGGACAGATACTTCCAAAAGGAATATTAGAGATTCCTAAGTTTGGATGTATAAATGTTCATGTTTCTTTACTTCCAAAATATAGAGGTGCGGCACCTATAAATTGGGTAATAATAAATGGTGAAGAAAAGACTGGTGTTACAACTATGTATATGGATGAAGGTCTAGATACTGGA
>M01808:26:000000000-A6KTK:1:1101:19201:1002
NCCAGTATCTAGACCTTCATCCATATACATAGTTGTAACACCAGTCTTTTCTTCACCATTTATTATTACCCAATTTATAGGTGCCGCACCTCTATATTTTGGAAGTAAAGAAACATGAACATTTATACATCCAAACTTAGGAATCTCTAATATTCCTTTTGGAAGTATCTGTCCAAAAGCTACAACTACTATTACATCTGGATTTAAAGATTTAATTGTATCTATAAATTCTTTATCTCTAGCCTTTACTGGTTGATATACAGGTATATTATTTTCAATTGCCAGTTCTTTTA
>M01808:26:000000000-A6KTK:1:1101:12506:1003
NAAAGATATTATTTTTAGCCCTGGTGTTGTACCTGCTGTTGCTATTTTAGTAAGAATATTAACTAATTCTAATGAAGGCGTGATAATTCAAAAGCCAGTGTATTACCCATTTGAAGCTAAGGTAAAGAGTAATAATAGGGAAGTTGTAAACAATCCTCTAATATATGAAAATGGGACTTATAGAATGGATTATGATGATTTGGAAGAAAAAGCTAAGTGTAGCAACAATAAAGTACTGATACTTTGTAGCCCTCACAATCCTGTTGGAAGAGTTTGGAGAGAAGATGAATTAAAAAAGGTT
>M01808:26:000000000-A6KTK:1:1101:12506:1003
NAGATTAAATGTTTTACTTGGAGCTATACATGTAACTATTTTATCCTTGTACTCTGGGCATAATGACTGTAAAGGAGTATGTTTAAATCCTTTTCTAATTAAATCAGAATGTATCTCATCAGCTATTATCCATAGGTCATATTTTTTACATATTTCTACAACCTTTTTTAATTCATCTTCTCTCCAAACTCTTCCAACAGGATTGTGAGGGCTACAAAGTATCAGTACTTTATTGTTGCTACACTTAGCTTTTTCTTCCAAATCATCATAATCCATTCTATAAGTCCCATTTTCATATATT
:
Get %GC content from reference FASTA file:
[uzi@quince-srv2 ~/linuxTutorial]$ bioawk -c fastx '{ print ">"$name; print gc($seq) }' data/Cdiff078.fa | less
>Cdiff078_C01
0.28096
>Cdiff078_C02
0.307669
>Cdiff078_C03
0.514628
>Cdiff078_C04
0.26898
>Cdiff078_C05
0.291059
>Cdiff078_C06
0.286006
>Cdiff078_C07
0.282794
>Cdiff078_C08
0.289484
:
Get the mean Phred quality score from a FASTQ file:
[uzi@quince-srv2 ~/linuxTutorial]$ bioawk -c fastx '{ print ">"$name; print meanqual($qual) }' data/M120_S2_L001_R1_001.fastq | less
>M01808:26:000000000-A6KTK:1:1101:19201:1002
37.3788
>M01808:26:000000000-A6KTK:1:1101:12506:1003
36.9867
>M01808:26:000000000-A6KTK:1:1101:19794:1003
37.1694
>M01808:26:000000000-A6KTK:1:1101:20543:1021
37.01
>M01808:26:000000000-A6KTK:1:1101:14616:1037
33.9133
>M01808:26:000000000-A6KTK:1:1101:10885:1044
35.9502
:
You want to see how many sequences are shorter (less than 1000bp?)
[uzi@quince-srv2 ~/linuxTutorial]$ bioawk -cfastx 'BEGIN{ s = 0} {if (length($seq) < 1000) s += 1} END {print "Shorter sequences", s}' data/Cdiff078.fa
Shorter sequences 12
You can count sequences very effectively with Bioawk, because NR now stores number of records:
[uzi@quince-srv2 ~/linuxTutorial]$ bioawk -cfastx 'END{print NR}' data/630_S4_L001_R1_001.fastq
329396
Further Reading
In the context of the exercises, it will be helpful if you could read through the following online tutorials, though it is not essential:
Bash tutorial (https://jack.logicalsystems.it/homepage/techinfo/Guida-Bash.txt)
Awk oneliners (http://www.pement.org/awk/awk1line.txt)
Sed oneliners (http://sed.sourceforge.net/sed1line.txt)
Perl oneliners (http://www.catonmat.net/download/perl1line.txt)
VI tutorial (http://www.nanocontact.cz/~trunec/education/unix/vi-tutor.txt)
You can also check my other one-liners specific to NGS data processing here:
Perl one-liners
Extracting information from GBK files
Bash one-liners for extracting enzyme information from annotated GBK files
Identifying duplicates in two FASTA files (awk)
Converting "Sample[TAB]Feature[TAB]Abundance" list to tab-delimited abundance table
Dereplicating Reads
Paired-end assembler
Subsampling FASTA and FASTQ files
Getting linkage information (reads that map to multiple contigs/genomes) from SAM files
Extracting subset of records from FASTA/FASTQ files based on exact/pattern matches of IDs
Spatial comparison of read qualities between different sequencing runs (Illumina paired-end reads)
Extracting 16S rRNA sequences from NCBI's locally installed nt database using blastdbcmd
Resolving NCBI Taxonomy using BioSQL
Generating abundance tables and trees from CREST and RDP classifiers
