6. 简单基因家族分析
2023.09.24
1. 任务背景
芝麻是一种油料作物.产油比其他作物高很多,这里以基因的背景来研究芝麻产油的原因.这里我们专门研究FAD4基因,它在油脂合成中也起到重要作用.我们对比不同作物的FAD4基因的拷贝数,研究它对产油的影响.
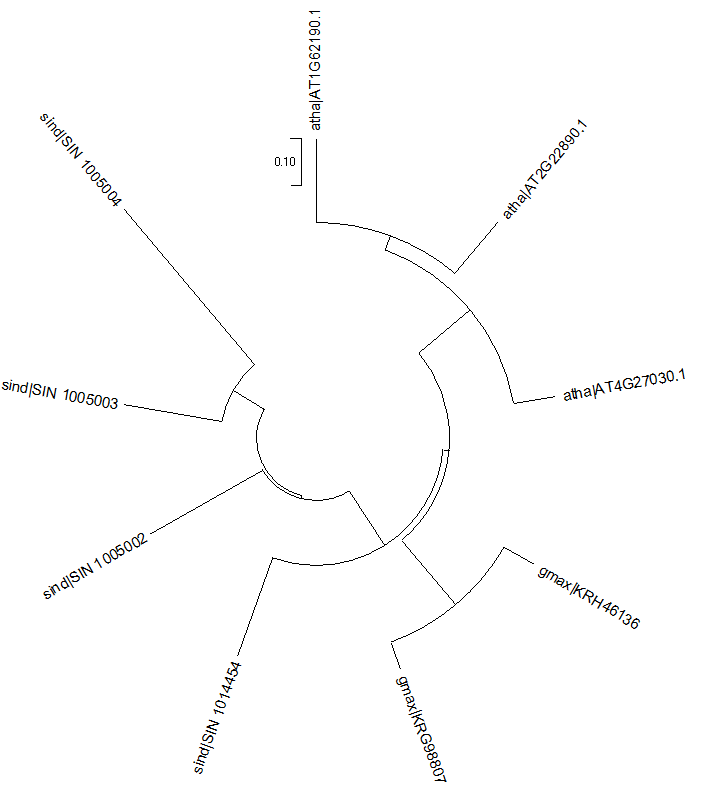
下图是我们要得出的结论,我们发现FAD4在拟南芥中有3个拷贝,在大豆中有2个拷贝,在芝麻中有5个拷贝,最后构建一个进化树.

但得出这个结论需要我们进行基因分析,我们可以发现拟南芥的基因有比较全的资料,因此找到拟南芥的FAD4基因,然后在大豆和芝麻中进行相似度分析,找到芝麻中比较相似的5个基因,对大豆同理.
2. 项目准备
首先就是需要建立项目路径,我们在workspace下建立项目文件夹FAD4.找到相似序列的工具是blast.BLAST(Basic Local Alignment Search Tool)是一套在蛋白质 数据库 或DNA数据库中进行相似性比较的分析工具.BLAST程序能迅速与公开数据库进行相似性序列比较.BLAST结果中的得分是一种对相似性的统计说明,more.
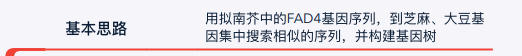
找到对应工具后,需要的就是找到工具的输入,也就是拟南芥的FAD4序列.这个序列正常来说,可以找蛋白序列,也可以是c
ds序列.可以看这个Click,简单来说cds是翻译成蛋白质的RNA序列,它没有内含子.
这是一个经常被人混淆的两个概念;CDS是Coding sequence的缩写,是指编码一段蛋白产物的序列,是与蛋白质密码子一一对应的序列,注意其与mRNA序列的差异;ORF是open reading frame的缩写,翻译成开放阅读框,是指从一个起始密码子开始到一个终止密码子结束的一段序列,但并不是所有读码框都能表达出蛋白产物;CDS必定是一个ORF,但也可能包括多个ORF,相反,每个ORF不一定都是CDS.
拟南芥的序列存放在atha_FAD4.fa文件中.用less指令查看如下图,这里使用的是蛋白序列(说是cds序列存在同义替换,没理解什么意思)
除了拟南芥的蛋白序列外,还需要大豆蛋白序列和芝麻的蛋白序列集合.
存储这些序列信息的文件是fasta文件.
- 第一行是由大于符号>打头的任意文字说明,主要为标记序列用.内容可以随意,但不能有重复,相当于身份识别信息
- 从第二行开始是序列本身,标准核苷酸符号或氨基酸单字母符号.通常核苷酸符号大小写均可,而氨基酸一般用大写字母.文件中和每一行都不要超过80个字符(通常60个字符).
3. 基因分析
我们确认思路:类比数据库查询,我们需要将拟南芥的蛋白序列为query,而大豆和芝麻的蛋白序列为database.蛋白对比使用的是blastp.按道理我们需要两次比对,但我们可以将大豆和芝麻的序列集合合并在一起(但必须保证ID不回复),查询一次即可.
# 当cat 指令后只接一个文件时,它会读取该文件内容,当后面接多个文件,会将他们拼在一起在读取
cat sind.fasta gmax.fasta > all.fasta
可以尝试使用blastp指令比对,但是会出现下面的问题,这是因为没有对db进行预处理.

makeblastdb可以对db进行数据库的处理,也就是使用makeblastdb创建自定义搜索库
## 构建数据库
makeblastdb -in genome.fasta -dbtype nucl -parse_seqids -out ./index
-
-in:构建数据库所用的序列文件。
-
-dbtype:数据库类型。构建的数据库是核苷酸数据库时,dbtype设置为nucl,数据库是氨基酸数据库时,dbtype设置为prot。
-
-out:数据库名称.
我们构建数据库后,会生成下图所示的几个文件,当我们使用blastp指令搜索的时候,就需要构建的文件.
![image]()
使用Blastp比对,输出文件格式可以随意.下面是用Blastn举得例子,blastp和它差不多.
## 将核苷酸序列比对至核苷酸数据库
blastn -query input.fa -db ./index -evalue 1e-6 -outfmt 6 -num_threads 6 -out out_file
-
-query:进行检索的序列。
-
-db:使用的数据库。
-
-evalue:设置输出结果中的e-value阈值。e-value低于1e-5就可认为序列具有较高的同源性。
# 蛋白序列比对代码
blastp -query atha_FAD4.fa -db all -out blast.out -evalue 1e-5
使用less指令查看输出结果.
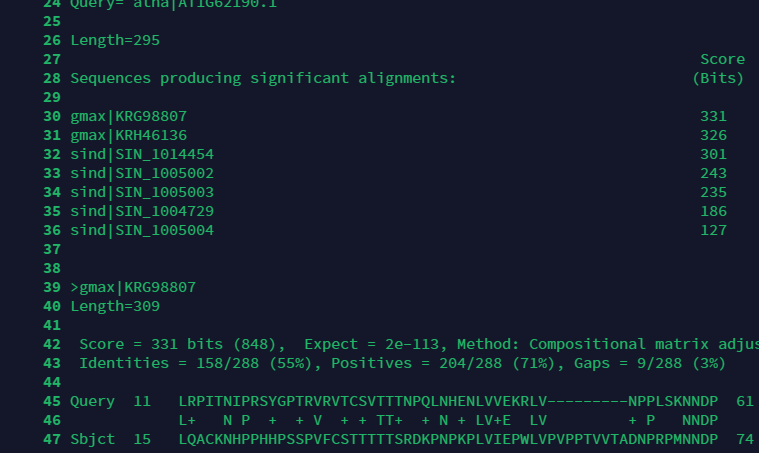
blast.out一开始将与它最相似的7条序列排列出来了,后续还列出了为什么如此详细.但这里只适用与少量的序列分析,如果我们需要一个精简版的,就需要调整指令.blastp还有选项如下:
-
-outfmt:输出文件的格式,一般设置为6。
-
-num_threads:线程数。
-
-out:输出文件。
![image]()
因此,调整一下指令就是:
blastp -query atha_FAD4.fa -db all -out blast.out -evalue 1e-5 -outfmt 7
接下来我们要对比对结果进行一个过滤.我们过滤自然要选择指标,一般是identity或者evalue.首先是去除注释行,这里我们选择grep指令.
less -SN blast.out | grep -v "#" > clean.out
# grep -v 表示只留下不匹配的行
# or ^#表示以#开始的行
grep -v "^#" blast.out | less -SN
去除了#号,我们考虑根据指标去除掉一些不是很匹配的行,这里我们考虑\(identity<50\)且\(evalue\)>\(e^{-10}\)的行去除.
grep -v "^#" blast.out | awk '$3>50' | awk '$11<1e-10' | less -SN
#也可以写成
grep -v "^#" blast.out | awk '$3>50 && $11<1e-10' | less -SN
#最后我们存到一个文件里
grep -v "^#" blast.out | awk '$3>50' | awk '$11<1e-10' | less -SN > blast.filtered.out
过滤后,剩下的都是符合要求的序列,此时我们将符合条件的序列ID提取出来,用于构建进化树(多序列比对).
- awk {print $2} blast.filtered.out 提取文件的第二列
此时可以将此句和上句合并,得到的输出如下图所示.
grep -v "^#" blast.out | awk '$3>50 && $11 < 1e-10{print $2}' | less -SN
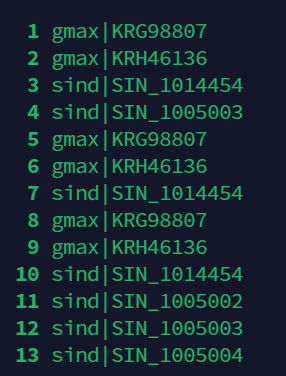
我们可以发现列表存在重复序列,如果我们要建树就必须将重复序列去掉.Linux用于去重的指令是\(uniq\).由于\(uniq\)命令只能对相邻行进行去重复操作,所以在进行去重前,先要对文本行进行排序,使重复行集中到一起。
grep -v "^#" blast.out | awk '$3>50 && $11 < 1e-10{print $2}' | sort | uniq > blast.list
上个步骤后,可以得到构建树需要的序列ID,但我们还需要序列本身,因此需在all.fasta里搜索.
这里需要使用的是seqtk软件.使用Conda或者Mamba安装后,我们需要使用seqtk搜索序列,使用方法可以参考这个Click.
seqtk subseq all.fasta blast.list > blast.fa
但是这样只是将大豆和芝麻的基因放在了\(blast.fa\)里,这里我们还需要放进拟南芥的基因.
cat atha_FAD4.fa >> blast.fa
blast.fa就是最终文件了,我们使用MEGA构建进化树.
4. blast 简单原理阐述
blast比对的算法是动态规划算法.其算法过程可简单描述为:
-
从两个序列中找出一些长度相等且可以形成无空位完全匹配的子序列,即序列片段对;
-
找出两个序列之间所有匹配程度超过一定值的序列片段对;
-
将得到的序列片段对根据给定的相似性阂值延伸,得到一定长度的相似性片段,称为高分值片段对。
Click了解更多.

 浙公网安备 33010602011771号
浙公网安备 33010602011771号