3. Linux 基础
1. Linux 版本
当今Linux有许多流行的版本.Centos可以看作Redhat的社区版

2. Linux 服务器
服务器对于不同用途也会有不同的配置.比如下面的塔式服务器就比较适合小公司.

比较生草的是,这次的课程使用的是提供的远程服务器,所以我们不用安装虚拟机了(.)
3. 总览
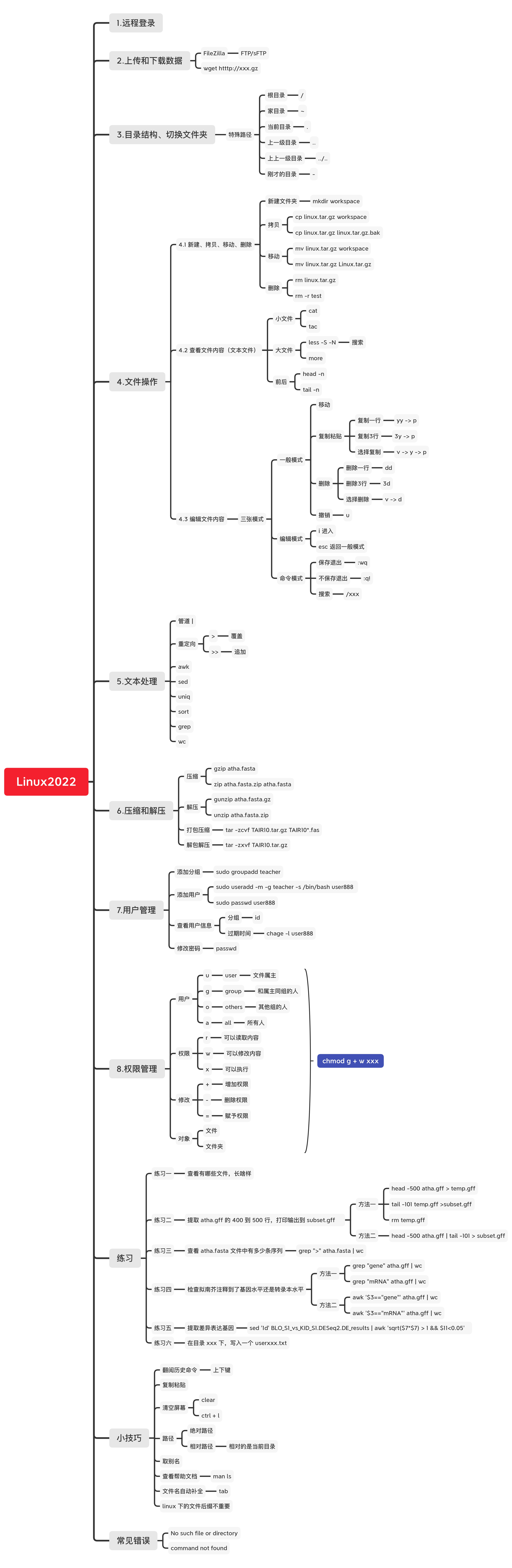
3.1 Linux 远程登陆
接下来是远程登陆Linux服务器,学校上课使用的是XShell,这里使用的是Mobaxterm.
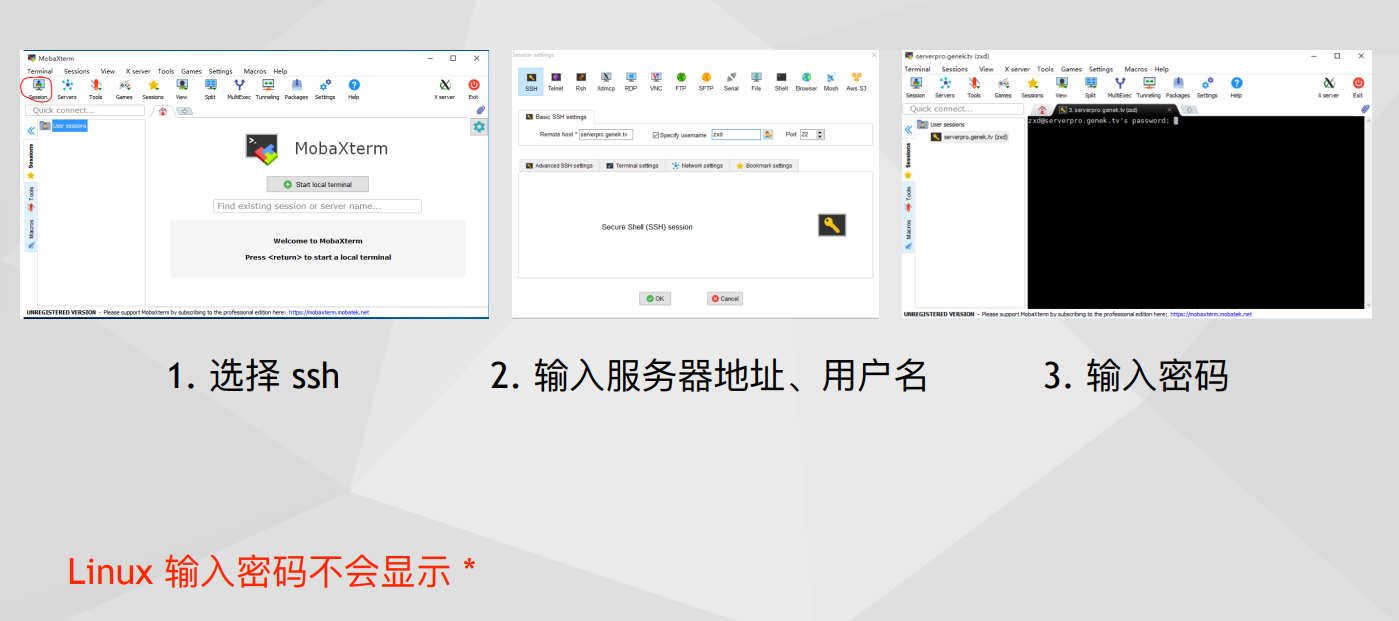
3.2 Linux 目录结构
虽然早就学过了,就当复习一遍吧....bin目录是用户可执行文件,etc目录是配置文件.home存储用户文件,默认用户只能在自己的家目录操作.
使用\(ls\)指令查看当前目录文件,\(-a\)参数显示全部信息.以.开头的文件表示隐藏文件.

如果要回到家目录,可以使用默认也可以~.
cd ~ or cd
#在刚才进入的目录与当前目录之间来回切换
cd -
# 下载 这个网址如果放在浏览器能有下载提示,就可以下载,如果没有下载提示就不能
wget 网址
3.3 上传与下载
FTP的默认端口是21,但是我们这次使用的协议是sFTP,默认端口是22.我们将网上能下载文件的网址通过Linux指令,让Linux服务器将文件下载到Linux的文件夹里.
这里吐槽一下,terminu的复制粘粘是ctrl+shift+c/v
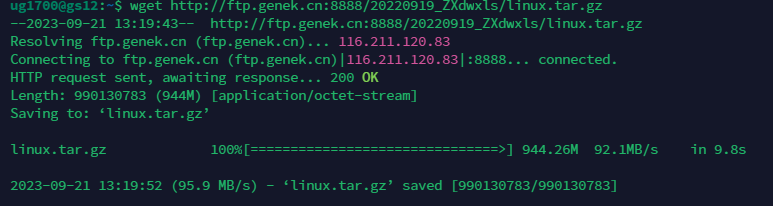
如果我们下载中途想杀死进程,就ctrl+c
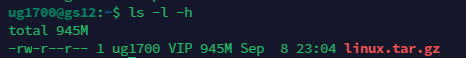
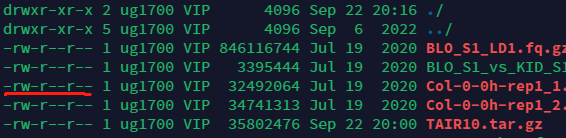
\(ls\ -l\)可以看到下载文件的详细信息.\(-rwxxxx\)是权限,1是硬链接数.VIP是用户分组.9901...表示文件大小,日期是文件最后的修改时间.

9901可能对我们来说难以理解,可以通过指令让他换算单位.
3.4 Linux 基本操作
这个看Linux的笔记总结更好,我这里就随便写点了.
3.4.1 Linux 文件操作
(1) 删除
rm 文件名
rm -r 文件夹名 # -r 表示循环递归
rm -f 文件名 # -f 强制删除
rm -r * 删除所有文件
rm -rf / #删除根目录
(2) 移动
cp 文件名 目标位置 #这其实是复制
rm 文件名
或者
mv 文件名 目标位置
mv 文件名 文件名 #这相当于重命名,如果是文件夹不需要加-r
(3) 删除
rm 文件名
rm
-
gz
gzip 文件路径 #请注意源文件会被删除
gunzip 文件路径 -
zip
解压:unzip FileName.zip
压缩:zip FileName.zip DirName(路径)
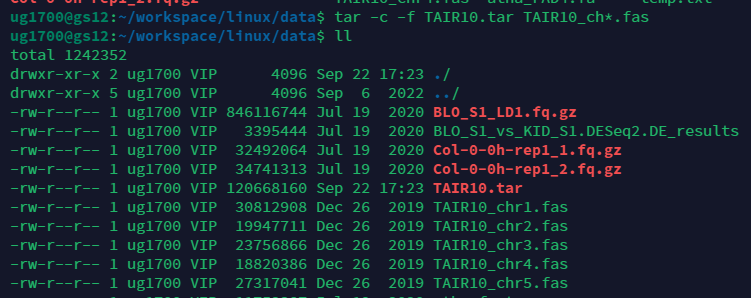
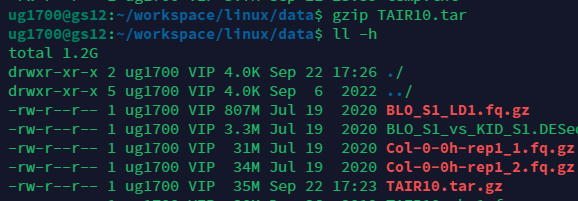
有时候我们需要打包多个文件,上面仅仅是压缩一个文件的例子.我们可以使用tar指令,压缩后文件后缀为tar.指定要压缩的文件可以使用通配符.但是请注意大小,这仅仅是打包并没有压缩.因此还有步骤,看下两张图的区别,变成了35M.
gzip TAIR10.tar
#请注意后缀,打包的后缀是tar,压缩后才是gz
但是tar也提供了顺便压缩的服务,使用-z即可
tar -zcf TAIR10.tar TAIR10_chr*.fas
#我们也可以加参数-v 可以显示压缩进度
tar -zcvf TAIR10.tar.gz TAIR10_chr*.fas
总结一下,-c是打包,-z是压缩,-v是显示进度,-f是指定操作打包后的名字,并且f选项后必须接变量名,所以顺序不能与前3个互换.
tar也可以解压.z选项识别格式,教程表明-z表示调用gzip压缩程序的功能.
tar -zxvf TAIR10.tar.gz
(5) 查看
linux下的文件格式不重要,不管是什么后缀,只要是文本文件都可以查看.但是为了格式统一,还是txt.
cat 文件路径
less 文件路径 # 这个适合用于查看大文件
less -S 文件路径 # 将一行的内容在一行显示
less -N 文件路径 # 显示行号
head 文件路径 #查看前几行
tail 文件路径 #查看后几行 默认皆为10行
head -n +x 文件路径 # 显示文件前x行的内容
tail -n +x 文件路径 # 显示文件x行到末尾的内容
tac 文件路径 # 从末尾行开始显示文件内容
more #与less类似 以一页一页的形式显示,更方便使用者逐页阅读,而最基本的指令就是按空白键(space)就往下一页显示,按 b 键就会往回(back)一页显示,而且还有搜寻字串的功能
查看的同时也可以搜索,使用less 查看文件后,先后/要查找的字符串就会自动找到在文件中的位置.
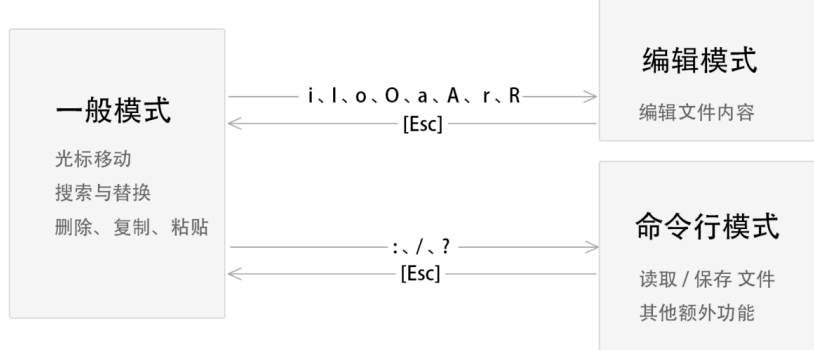
(6) 编辑
在一般模式
- w: 保持
- q: 退出
- wq: 保存退出
- q!: 不保存退出
- yy: 复制
- p: 粘贴
- v: 转换到可视模式,然后上下移动选中行,可按y复制
- dd:删除; x dd --> 删除x行
- u: 撤销
- /:搜索
![image]()
在用vim打开一个文件时,会产生一个cmd.swap文件,用于保存数据:
1、当编辑中的文件非正常关闭时,可用此.swp文件来恢复(非正常关闭时,不会被删除,所以提示存在.swap文件);
2、当编辑文件正常关闭时,此.swp文件会被删除;
3.4.2 用户管理
(1) 查看当前用户信息

(2) 创建分组
sudo groupadd teacher
#以管理员的身份运行
(3) 创建用户
useradd [选项] 用户名

(4) 设置密码
passwd 用户名
(5) 设置账号过期时间
chage [选项] 用户名
-m:密码可更改的最小天数。为零时代表任何时候都可以更改密码。
-M:密码保持有效的最大天数。
-w:用户密码到期前,提前收到警告信息的天数。
-E:帐号到期的日期。过了这天,此帐号将不可用。
-d:上一次更改的日期。
-I:停滞时期。如果一个密码已过期这些天,那么此帐号将不可用。
-l:例出当前的设置。由非特权用户来确定他们的密码或帐号何时过期。
3.4.3 权限管理
权限将用户分为三类,用户对文件的操作也可以分为三类.当我们使用\(ll\)查看具体文件信息时,可以看到三类用户对文件的可执行权限.
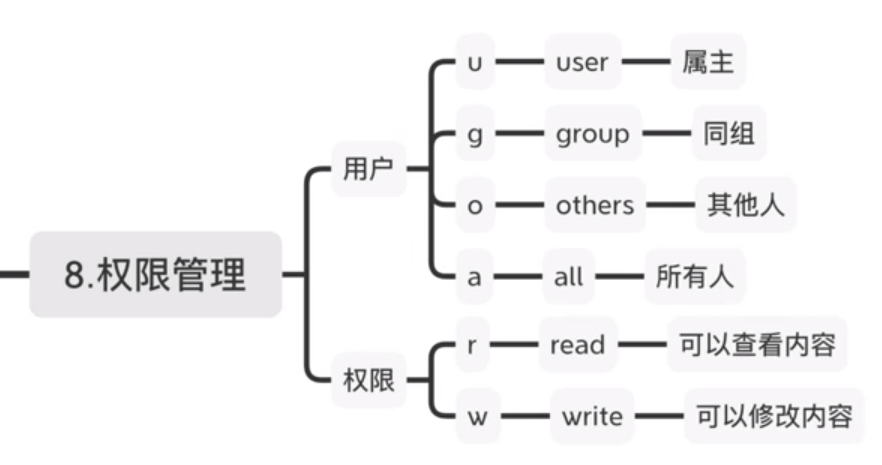
请注意画红线处,第一个字符表示文件类型,接下来9个字符表示\(ugo\)三类用户的权限.
使用chmod修改权限.
chmod [-cfvR] [--help] [--version] mode file...

稍微解释一下文件夹的权限.能看见文件夹的内容,就是文件夹的r权限,能在里面创建(or 删除)文件,就是w权限,能进入文件夹,就是x权限.
请注意,我们修改文件夹的权限,如果不使用-R,就只是修改当前文件夹的权限,下一级文件夹的权限并没有修改
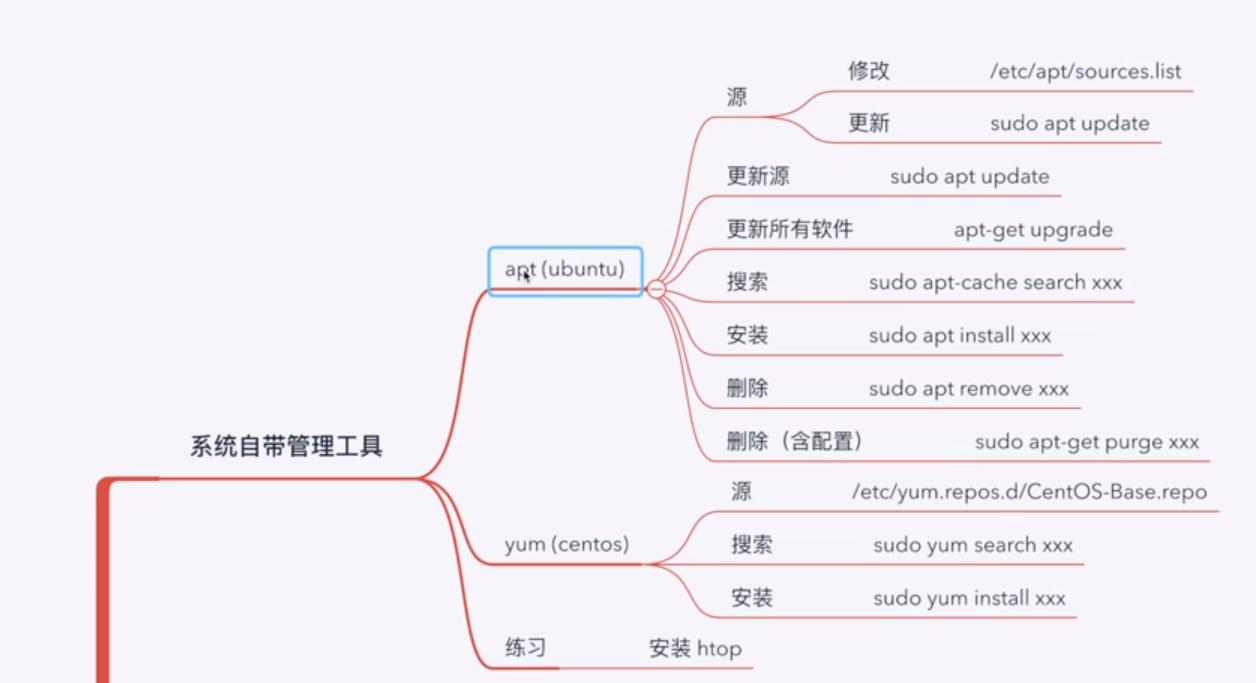
3.4.4 软件管理
linux安装软件主要是下面几个指令,请注意,如果我们要修改sources文件的管道,就要先知道Ubuntu的版本,然后在使用匹配的管道.
(1) htop
使用htop可以观察系统全部进程情况,上半部分是CPU使用情况.
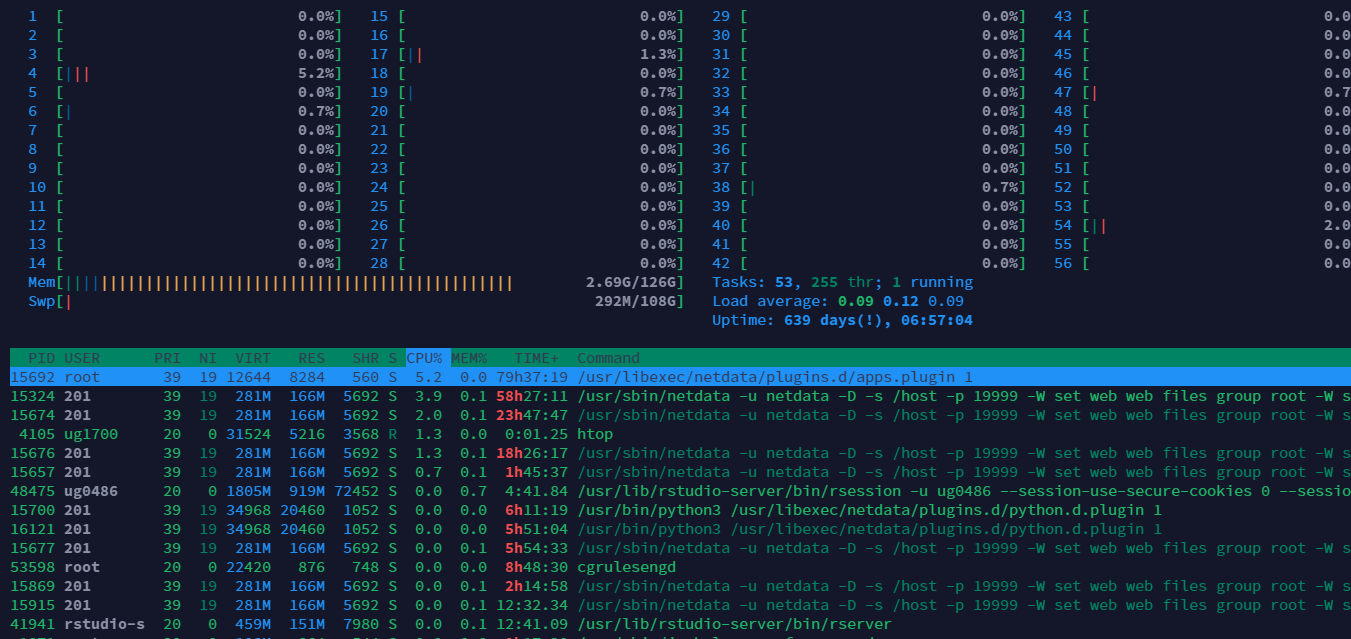
使用which htop,查看软件的安装路径.which -a xxx表示去所有环境变量下找.
(2) fastp
按照这个Link安装.作者给Linux用户使用了预编译的版本(预先编译成机器语言).
当我们使用这个指令时,每次都需要指定指令的路径,如果不想指定,就需要配置环境变量.$PATH是Linux存储环境变量的路径.下面是将fastp的路径加入到PATH的指令:
#$PATH表示之前未赋值的PATH,=表示新加上路径然后赋值
export PATH=/home/ug1700/software:$PATH
#但是请注意, 上句代码只对当前会话生效
如果我们要对全部的会话自动生效,需要在/home/用户目录修改配置文件
vim .bashrc
再将export PATH=/home/ug1700/software:$PATH添加到文件末尾
#原理是当登陆到服务器上时,会先将.bashrc运行一遍,所以当我们重新登陆后,就可以任意调用
#source .bashrc 也可以强制执行一遍
3.4.5 脚本管理
当我们需要执行大量相同或相似的命令时,就需要Linux脚本.我们在脚本里编写命令,通过sh xx.sh调用\(xx.sh\)执行.比如下面这个:
# 记录完成这个项目使用的指令
makeblastdb -in all.fasta -dbtype prot -out all
blastp -query atha_FAD4.fa -db all -out blast.out -evalue 1e-5 -outfmt 7
grep -v "^#" blast.out | awk '$3>45 && $11 < 1e-10{print $2}' | sort | uniq > blast.list
seqtk subseq all.fasta blast.list > blast.fa
cat atha_FAD4.fa >> blast.fa
但是如果脚本我们需要变量,就可以稍微改一下,请注意$表示引用:
# 记录完成这个项目使用的指令
query=atha_FAD4.fa
makeblastdb -in all.fasta -dbtype prot -out all
blastp -query $query -db all -out blast.out -evalue 1e-5 -outfmt 7
grep -v "^#" blast.out | awk '$3>45 && $11 < 1e-10{print $2}' | sort | uniq > blast.list
seqtk subseq all.fasta blast.list > blast.fa
cat $query >> blast.fa
我们也可以让脚本允许命令行传参,$1表示从命令行传递进来的第一个参数:
# 记录完成这个项目使用的指令
query=$1
makeblastdb -in all.fasta -dbtype prot -out all
blastp -query $query -db all -out blast.out -evalue 1e-5 -outfmt 7
grep -v "^#" blast.out | awk '$3>45 && $11 < 1e-10{print $2}' | sort | uniq > blast.list
seqtk subseq all.fasta blast.list > blast.fa
cat $query >> blast.fa
我们调用sh脚本时,一般是\(sh\ xx.sh\),我们也可以直接打\(xx\)不打后缀名.\(sh\ xx.sh\)表示xx.sh用sh来解释.如果我们在脚本文件的首行加上下列代码,就可以直接使用./xx.sh调用.但是记得要给\(xx.sh\)增加可执行权限.这时就可以把文件名的sh也给去掉了mv run.sh run.如果我们将run加入到环境变量,就可以在任意路径下运行.
#!/bin/bash
3.4.6 其他操作
(1) 起别名
我们经常使用less -S命令,如果嫌它太长了,可以给指令起别名,当然我们再次登录这个指令就不在生效.解决方法就是配置环境变量,请参考这个总结的环境变量顺序与生效范围Click.
alias xx='less -S'
#xx就可以代表less -S
4. 练习
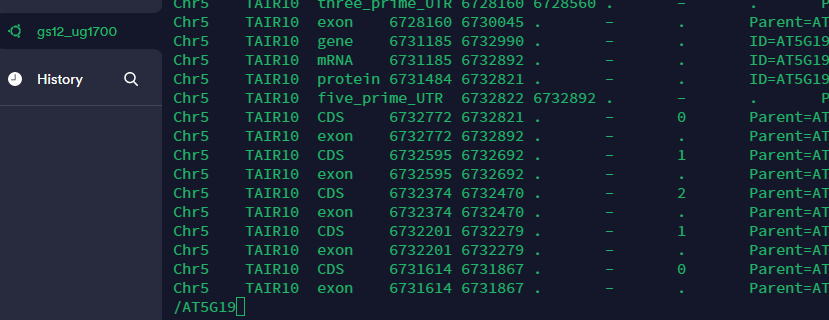
4.1 提取 atha.gff 的 400 到 500 行,打印输出到 subset.gff
(1) 方法一:先取atha.gff的前500行放在一个文件里,再从新文件取后100行.
- 重定向指令 >
将>前的内容覆盖到文本
head -n +500 atha.gff > temp.gff
tail -n +101 temp.gff > subset.gff #如果写100的话,就是101行,而非100行
rm temp.gff
(2) 方法二:其实可以简写为一行,|表示管道.后的参数是前的结果.
head -n +500 atha.gff | tail -n +101 > subset.gff
我们可以记一下这些符号命令

4.2 查看 atha.fasta ⽂件中有多少条序列
利用wc指令我们可以计算文件的Byte数、字数、或是列数,若不指定文件名称、或是所给予的文件名为"-",则wc指令会从标准输入设备读取数据.在默认的情况下,wc将计算指定文件的行数、单词数(不包含符号),以及字节数(字符).

我们要计算有多少序列,需要计算有多少行具有>.需要引入grep指令.grep (global regular expression) 命令用于查找文件里符合条件的字符串或正则表达式
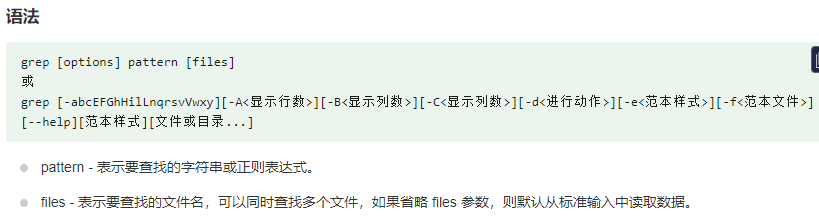
grep ">" atha.fasta | wc
# 后面的指令的参数是前面的结果
4.3 检测拟南芥有多少条转录本
只计算mRNA,用grep即可
#计算mRNA
grep "mRNA" atha.gff | wc
#计算基因
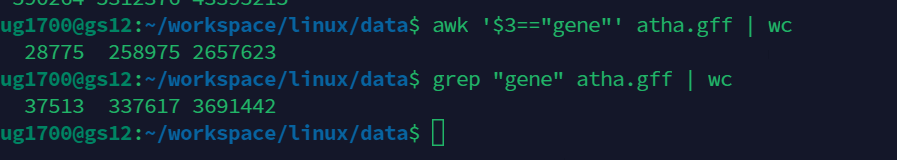
grep "gene" atha.gff | wc
基因注释是弄清楚基因组有多少条基因,每个基因的起始点和终止点在哪里.这就是基因的结构注释.
当mRNA比gene多,就注释到了转录组水平,否则就是gene水平.
上面是一个比较简单的做法.但是我们通yige过观察gff文件可以发现,在非第三列也存在gene字符串,因此每行抓取gene字符串并不是一个比较好的做法.
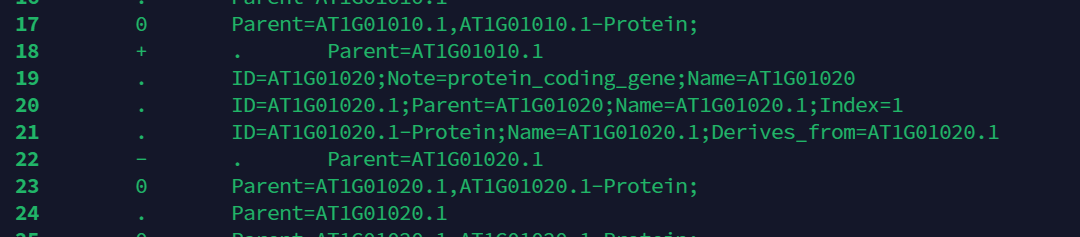
这里介绍一个指令awk.awk 是一种编程语言,用于在linux/unix下对文本和数据进行处理。数据可以来自标准输入(stdin)、一个或多个文件,或其它命令的输出.

基本命令格式如下:
awk '条件1 {动作 1} 条件 2 {动作 2} …' 文件名
使用方法有点复杂,建议看这个Click.
下图是搜索atha.gff的第三列(默认以空格为分隔符)==gene的行数.
4.4 提取差异表达基因
在BLO_S1_vs_KID_S1.DESeq2.DE_results文件中,查看文件如下图:
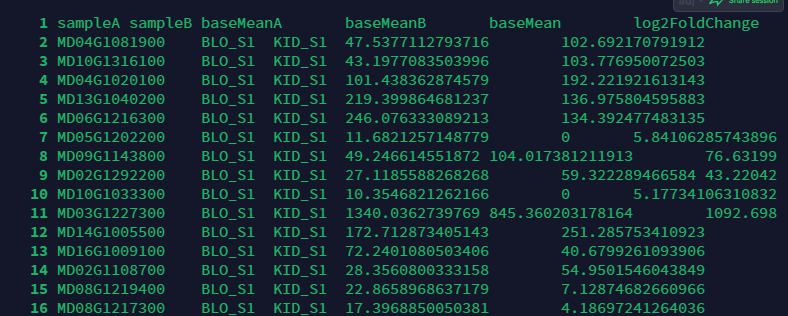
每一行都代表一个基因,当log2FoldChange<0时,表示表达量下降了,如果log2FoldChange>0,表示表达量上调了.如果是差异表达基因,则这个值的绝对值>1,且padj这列绝对值<0.05
#先不考虑绝对值的情况,且不考虑表头
awk '$7>1 && $11<0.05' BLO_S1_vs_KID_S1.DESeq2.DE_results | less -SN
但实际上我们要去掉表头.这里引入一个指令叫sed.sed 命令的作用是利用脚本来处理文本文件.使用方法:
[root@localhost ~]# sed [选项] [脚本命令] 文件名
可以参考一下这个教程Click.
去掉表头的指令是:
sed '1d' BLO_S1_vs_KID_S1.DESeq2.DE_results > temp.DE_results
接下来是取绝对值的指令,Python里是abs,很遗憾awk用不了,但是可以使用sqrt函数.
awk 'sqrt($7*$7)>1' temp.DE_results | less -SN
条件合并就是
awk 'sqrt($7*$7)>1 && sqrt($11*$11)<0.05' temp.DE_results | less -SN
#或者上面指令全部缩写一句
sed '1d' BLO_S1_vs_KID_S1.DESeq2.DE_results | awk 'sqrt($7*$7)>1 && sqrt($11*$11)<0.05' | less -SN
建议uniq和sort都学学.

 浙公网安备 33010602011771号
浙公网安备 33010602011771号