目的
对于变异检测,通常每个样本都可以检测出许多变异,比如一个正常的全外可以检测 4w+ 个小变异(snv + indel), 而通常样本量多时,就有必要将所有检出的变异收集起来,用于后续的数据挖掘、变异查询工作。
数据挖掘可以是分析出罕见变异(数据库内频率非常低的变异),也可以是利用其人群很高的变异,排除非疾病相关的变异,亦或是实验、分析流程缺陷造成的假阳性变异(通常同样的实验、分析流程造成的假阳性变异比较一致)
逻辑
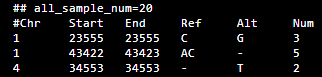
最简单的突变频率数据库构建方式,是按变异为行,储存其 "染色体编号,变异起止位置,ref碱基,alt碱基,变异出现次数", 当然还需要一个地方记录样本总数量才能得知在群体内的突变频率,数据库示例如下:
升级
简单形式的数据库(暂且将这个数据库记为 A),A 直观的记录着变异在群体内的发生的次数,但不足之处挺多的。
基因型
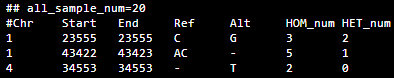
A 的一个问题是变异基因型没有记录,如果物种是人,纯杂合影响的疾病的程度就可能存在差别了。因此需要将 num 字段拆分为 HOM_num 和 HET_num 字段, A 可以改为如下形式(暂且记为 B):
样本追踪
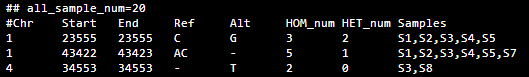
A、B 相类似,都没有记录这个变异存在于哪些样本中。增加 Samples 字段,记录样本覆盖这个变异的样本编号,因此可以改进数据库为如下形式(暂且记为 C):
探针覆盖
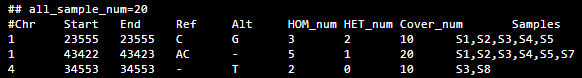
假如上个与使用的是 IDT 的全外探针检测了 a1 个样本,这个月打算用 NAD 的探针预期检测 a2 个样本,而变异位点 v1 只被 IDT 探针覆盖到,如果按数据库 C 的记录形式的话,v1 的群体频率计算公式为: ( Hom_num[v1] + HET_num[v1] ) / (a1 + a2), 而 v1 并没有被 NAD 探针覆盖到,正确的计算公式应为: ( Hom_num[v1] + HET_num[v1] ) / a1
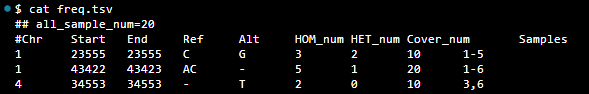
增加 Cover_num 字段,记录变异被覆盖的次数,如 “1:23555-23555 C->G HOM" 变异原群体频率为 15%,现在正确的群体频率为 30%。因此改进数据库为如下形式(暂且记为 D):
储存空间
一般样本编号可能有 10 个左右的字符。如果检测的样本量在万以上的级别,那么 Samples 字段最长可以达到 10w+ 个字符, 如果再加上总的变异数量万以上的级别,那可能光 Samples 列就有上亿个字符,无论是文本还是 mysql 等储存方式,都是比较大的负担。
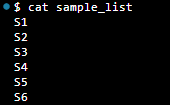
可以选择一个映射关系,将样本编号更换为索引号,节省记录的字符数量。如,将样本编号单独存放在一个样本列表文件内,数据库内以样本所在行号表示样本。而数据行号可以根据其顺序,进行简写、合并,如 “1,2,3,4,5,6” 可以简写为 “1-6” ,表示 sample_list 中第一到第六个样本都检测到了这个变异。
数据库改进示例如下(暂且记为 E):
其他可优化项
加速 | 归档
(如果是使用 mysql 之类的数据库操作,应该没有这个问题)
拿人的全外样本来说,常规一个样本检测 4w+ 个变异,但是样本与样本之间可能存在数千个各自特有的变异,因此数据库内存放的变异数量可能是 100w+ ,而每次更新的时候仅需更新其中 4w+ 个变异记录即可。
方案一:
- 利用
bedtools筛选变异列表一致的数据库变异记录 v1,不一致的变异列表 d2。 - 调整 v1 记录信息为 d1。
- 合并 d1 和 d2,排序。
方案二:
数据库分档,比如数据库一份只存 1000 个样本,多余的样本重新生成一份 E 结构,后续访问、查询方面需要注意多份文件的问题。
当然应该还会有更好更实用的方式做数据库的更新~
优先级
一个样本可能被分析很多次,也可能被内部使用不同的探针进行实验,此时就需要一套规则确定新上传的样本数据与数据库内该样本数据保留哪一份。如果是覆盖旧的,旧需要先清楚数据库内已存在的该样本所有变异,然后再重新上传该样本新的变异。
并行
当需要访问、使用数据库内的数据时,可以根据需要采用多线程加速。如一批 12 个样本需要对它们所有的变异进行群体突变频率注释,各样本都可以同时去访问数据库,任何两个变异也都可以同时计算群体突变频率,以此可加快数据使用效率。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号