08基因共表达分析
一、基因共表达数据准备
将要分析的基因放到gene.txt
参考https://www.cnblogs.com/lzryhc/articles/17826180.html,将转录组矩阵作为输入文件
panCancer23.geneCor.R
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("limma")
#引用包
library(limma)
gene="EPHA1" #基因名称,改成自己研究的基因
#setwd("D:\\biowolf\\panCancer\\23.geneCor") #设置工作目录
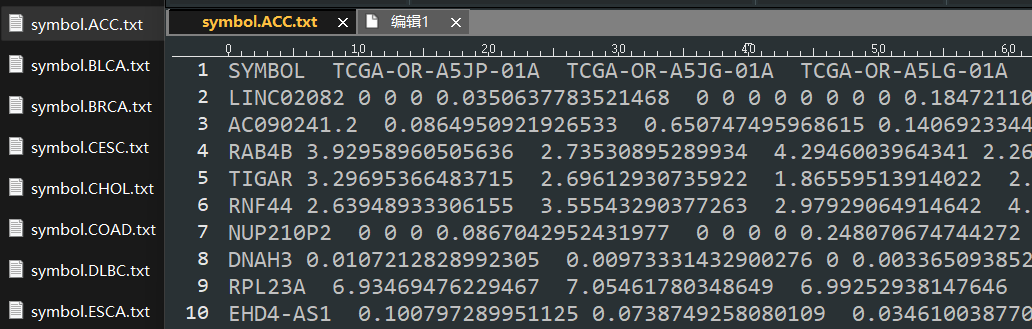
geneRT=read.table("gene.txt",sep="\t",header=F) #读取基因表达文件
files=dir()
files=grep("^symbol.",files,value=T)
#按肿瘤类型循环进行相关性检验
outTab=data.frame()
corTab=data.frame()
sameGenes=c()
for(i in files){
#读取表达文件
CancerType=gsub("^symbol\\.|\\.txt$","",i)
rt=read.table(i, header=T,sep="\t",check.names=F)
#如果一个基因占了多行,取均值
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
#去除正常样品
group=sapply(strsplit(colnames(data),"\\-"),"[",4)
group=sapply(strsplit(group,""),"[",1)
group=gsub("2","1",group)
data=data[,group==0]
#提取gene.txt里面基因的表达量
sameGenes=intersect(as.vector(geneRT[,1]),row.names(data))
data=data[c(sameGenes,gene),]
#对基因进行循环
x=as.numeric(data[gene,])
outVector=data.frame(CancerType)
corVector=data.frame(CancerType)
for(j in sameGenes){
y=as.numeric(data[j,])
if(sd(y)>0.01){
corT=cor.test(x,y)
cor=corT$estimate
pValue=corT$p.value
outVector=cbind(outVector,pValue)
corVector=cbind(corVector,cor)
}
else{
outVector=cbind(outVector,pValue=1)
corVector=cbind(corVector,cor=0)
}
}
outTab=rbind(outTab,outVector)
corTab=rbind(corTab,corVector)
}
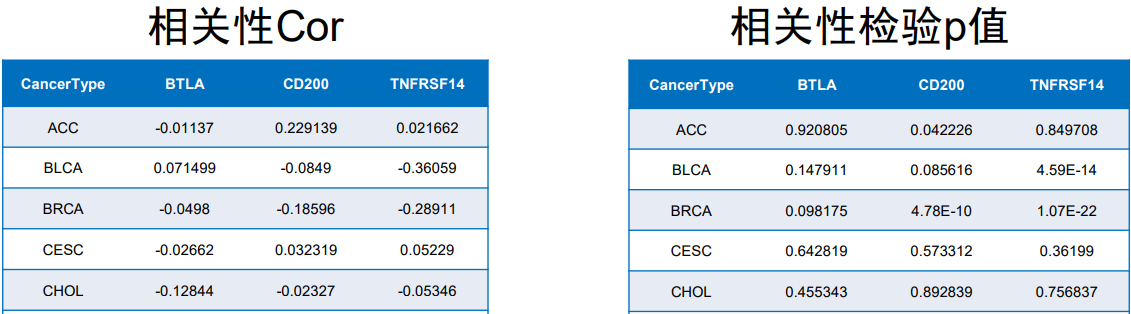
#输出相关性检验p值结果
colnames(outTab)=c("CancerType",sameGenes)
write.table(outTab,file="geneCor.pvalue.txt",sep="\t",row.names=F,quote=F)
#输出相关性结果
colnames(corTab)=c("CancerType",sameGenes)
write.table(corTab,file="geneCor.cor.txt",sep="\t",row.names=F,quote=F)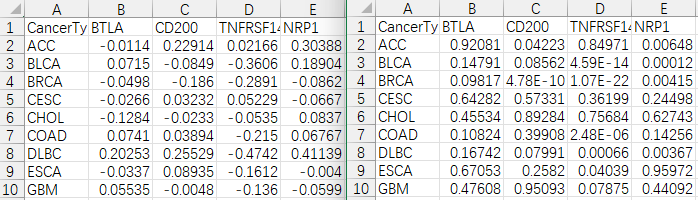
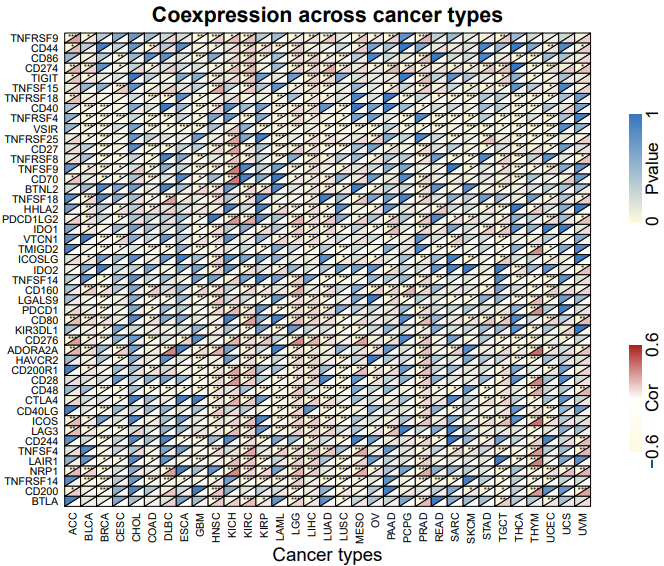
二、共表达热图
heatmap.R
#install.packages("reshape2")
#install.packages("RColorBrewer")
library(reshape2)
library(RColorBrewer)
options(stringsAsFactors = F)
#setwd("D:\\biowolf\\panCancer\\24.heatmap") #设置工作目录
up <- read.table("geneCor.pvalue.txt",sep = "\t",check.names = F,header = T,row.names=1) #读取左上角的数据
dn <- read.table("geneCor.cor.txt",sep = "\t",check.names = F,header = T,row.names=1) #读取右下角数据
dn=t(dn)
up=t(up)
#设置颜色
colVector=c("#AB221F","#3878C1","#FFFADD")
#行名和列名
gene.level <- as.character(rownames(dn))
cancer.level <- as.character(colnames(dn))
#把行转为列
dn.long <- setNames(melt(dn), c('Gene', 'Cancer', 'Frequency'))
dn.long$Categrory <- "DN"
up.long <- setNames(melt(up), c('Gene', 'Cancer', 'Frequency'))
up.long$Categrory <- "UP"
#右下角颜色
dn.long$range <- cut(dn.long$Frequency,
breaks = seq(floor(min(dn.long$Frequency)),
ceiling(max(dn.long$Frequency)),0.01))
rangeMat1 <- levels(dn.long$range) # 提出分割区间
rbPal1 <- colorRampPalette(colors = c(colVector[3],"white",colVector[1]))
col.vec1 <- rbPal1(length(rangeMat1)); names(col.vec1) <- rangeMat1
dn.long$color <- col.vec1[as.character(dn.long$range)]
#左上角颜色
up.long$range <- cut(up.long$Frequency, breaks = seq(floor(min(up.long$Frequency)),ceiling(max(up.long$Frequency)),0.01))
rangeMat2 <- levels(up.long$range)
rbPal2 <- colorRampPalette(colors = c(colVector[3],colVector[2]))
col.vec2 <- rbPal2(length(rangeMat2)); names(col.vec2) <- rangeMat2
up.long$color <- col.vec2[as.character(up.long$range)]
#合并右下角和左上角
heatmat <- rbind.data.frame(dn.long,up.long) #汇总热图矩阵
pdf("heatmap.pdf",width = 7,height = 6)
layout(mat=matrix(c(1,0,1,2,1,0,1,3,1,0),5,2,byrow=T),widths=c(length(cancer.level),2))
#热图绘制区域
par(bty="n", mgp = c(2,0.5,0), mar = c(5.1, 5.5, 3, 3),tcl=-.25,xpd = T)
x=as.numeric(factor(heatmat$Cancer,levels = cancer.level))
y=as.numeric(factor(heatmat$Gene,levels = gene.level))
#创建空白画布
plot(1,xlim=c(1,length(unique(x))+1),ylim=c(1,length(unique(y))+1),
xaxs="i", yaxs="i",xaxt="n",yaxt="n",
type="n",bty="n",xlab="",ylab="",
main = "Coexpression across cancer types",cex.main=2)
#填充颜色
for(i in 1:nrow(heatmat)) {
if(heatmat$Categrory[i]=="DN") polygon(x[i]+c(0,1,1),y[i]+c(0,0,1),col=heatmat$color[i]) #填充左上角
if(heatmat$Categrory[i]=="UP") {
polygon(x[i]+c(0,1,0),y[i]+c(0,1,1),col=heatmat$color[i]) #填充右下角
if(heatmat$Frequency[i]<0.001){
text(x[i]+0.5,y[i]+0.8,"***",cex=0.8)
}else if(heatmat$Frequency[i]<0.01){
text(x[i]+0.5,y[i]+0.8,"**",cex=0.8)
}else if(heatmat$Frequency[i]<0.05){
text(x[i]+0.5,y[i]+0.8,"*",cex=0.8)
}
}
}
#基因名和癌症名
axis(1,at = sort(unique(x)) + 0.5,labels = cancer.level,lty = 0,las = 2) #添加x轴坐标
axis(2,at = sort(unique(y)) + 0.5,labels = gene.level,lty = 0,las = 1) #添加y轴坐标
mtext("Cancer types",side = 1,line = 3.5,cex=1.2) #x轴名称
#绘制图例
par(mar=c(0,0,0,2),xpd = T,cex.axis=1.6)
barplot(rep(1,length(col.vec2)),border = NA, space = 0,ylab="",xlab="",ylim=c(1,length(col.vec2)),horiz=TRUE,
axes = F, col=col.vec2) # Loss
axis(4,at=c(1,ceiling(length(col.vec2)/2),length(col.vec2)),c(round(min(up),1),'Pvalue',round(max(up),1)),tick=FALSE)
par(mar=c(0,0,0,2),xpd = T,cex.axis=1.6)
barplot(rep(1,length(col.vec1)),border = NA, space = 0,ylab="",xlab="",ylim=c(1,length(col.vec1)),horiz=TRUE,
axes = F, col=col.vec1) # Gain
axis(4,at=c(1,ceiling(length(col.vec1)/2),length(col.vec1)),c(round(min(dn),1),'Cor',round(max(dn),1)),tick=FALSE)
dev.off()