08肿瘤微环境相关性分析
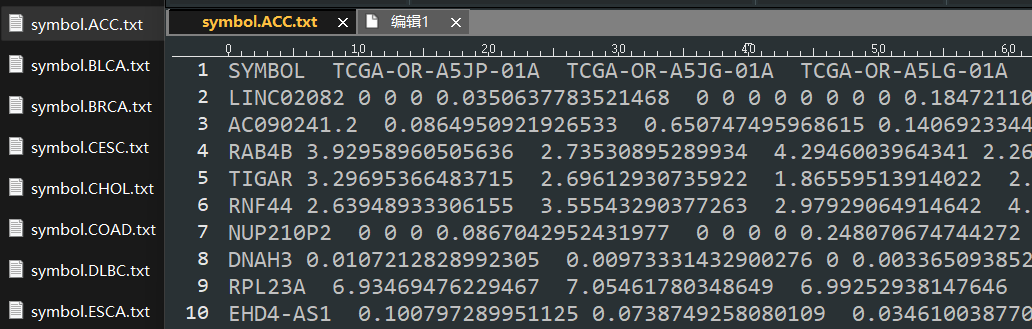
一、准备输入文件
将转录组矩阵作为输入文件
二、免疫环境评分
panGenes14.estimate.R
#library(utils)
#rforge <- "http://r-forge.r-project.org"
#install.packages("estimate", repos=rforge, dependencies=TRUE)
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("limma")
#引用包
library(limma)
library(estimate)
#setwd("D:\\biowolf\\panGenes\\14.estimate") #设置工作目录
#读取目录下的文件
files=dir()
files=grep("^symbol.",files,value=T)
outTab=data.frame()
for(i in files){
#读取文件
CancerType=gsub("symbol\\.|\\.txt","",i)
#CancerType="ACC"
rt=read.table(i,sep="\t",header=T,check.names=F)
#rt=read.table("symbol.ACC.txt",sep="\t",header=T,check.names=F)
#如果一个基因占了多行,取均值
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
#删除正常,只保留肿瘤样品
group=sapply(strsplit(colnames(data),"\\-"),"[",4)
group=sapply(strsplit(group,""),"[",1)
group=gsub("2","1",group)
data=data[,group==0]
out=data[rowMeans(data)>0,]
out=rbind(ID=colnames(out),out)
#输出整理后的矩阵文件
write.table(out,file="uniq.symbol.txt",sep="\t",quote=F,col.names=F)
#运行estimate包
uniqFile="uniq.symbol.txt"
inputDs="commonGenes.gct"
outputDs="estimateScore.gct"
filterCommonGenes(input.f=uniqFile, output.f=inputDs, id="GeneSymbol")
estimateScore(input.ds =inputDs ,output.ds=outputDs)
#输出每个样品的打分
scores=read.table("estimateScore.gct",skip = 2,header = T)
rownames(scores)=scores[,1]
scores=t(scores[,3:ncol(scores)])
rownames(scores)=gsub("\\.","\\-",rownames(scores))
outTab=rbind(outTab,cbind(scores,CancerType))
file.remove(uniqFile)
file.remove(inputDs)
file.remove(outputDs)
}
out=cbind(ID=row.names(outTab),outTab)
write.table(out,file="estimateScores.txt",sep="\t",quote=F,row.names=F)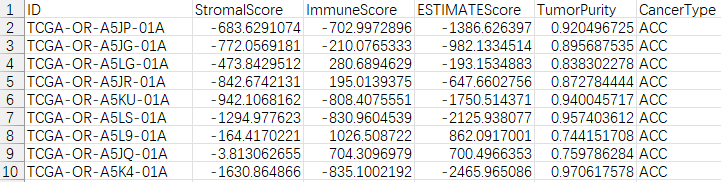
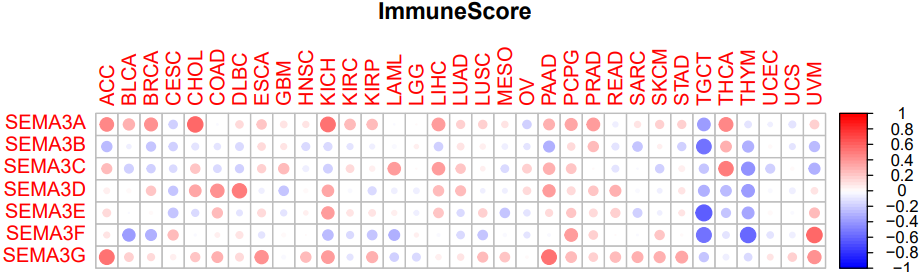
三、肿瘤微环境相关性StromalScore和ImmuneScore
panGenes15.estimateCor.R
#install.packages("corrplot")
library(corrplot) #引用包
expFile="panGeneExp.txt" #表达输入文件
scoreFile="estimateScores.txt" #免疫微环境结果文件
scoreType="StromalScore" #打分类型
#scoreType="ImmuneScore" #打分类型
#setwd("D:\\biowolf\\panGenes\\15.estimateCor") #设置工作目录
#读取表达文件
exp=read.table(expFile, header=T,sep="\t",row.names=1,check.names=F)
exp=exp[(exp[,"Type"]=="Tumor"),]
#读取肿瘤微环境文件
TME=read.table(scoreFile, header=T,sep="\t",row.names=1,check.names=F)
#样品取交集
sameSample=intersect(row.names(TME),row.names(exp))
TME=TME[sameSample,]
exp=exp[sameSample,]
#相关性检验
outTab=data.frame()
pTab=data.frame()
#按肿瘤类型循环
for(i in levels(factor(exp[,"CancerType"]))){
exp1=exp[(exp[,"CancerType"]==i),]
TME1=TME[(TME[,"CancerType"]==i),]
x=as.numeric(TME1[,scoreType])
pVector=data.frame(i)
outVector=data.frame(i)
#按基因循环
genes=colnames(exp1)[1:(ncol(exp1)-2)]
for(j in genes){
y=as.numeric(exp1[,j])
corT=cor.test(x,y,method="spearman")
cor=corT$estimate
pValue=corT$p.value
pVector=cbind(pVector,pValue)
outVector=cbind(outVector,cor)
}
pTab=rbind(pTab,pVector)
outTab=rbind(outTab,outVector)
}
#输出相关性的表格
colNames=c("CancerType",colnames(exp1)[1:(ncol(exp1)-2)])
colnames(outTab)=colNames
write.table(outTab,file="estimateCor.cor.txt",sep="\t",row.names=F,quote=F)
#输出相关性检验p值的表格
colnames(pTab)=colNames
write.table(pTab,file="estimateCor.pval.txt",sep="\t",row.names=F,quote=F)
#绘制微环境相关性图形
pdf(file=paste0(scoreType,".pdf"),height=5,width=8)
row.names(outTab)=outTab[,1]
outTab=outTab[,-1]
corrplot(corr=as.matrix(t(outTab)),
title=paste0("\n\n\n\n",scoreType),
col=colorRampPalette(c("blue", "white", "red"))(50))
dev.off()
四、肿瘤干细胞相关性(RNAss)
需要使用StemnessScores_RNAexp_20170127.2.tsv
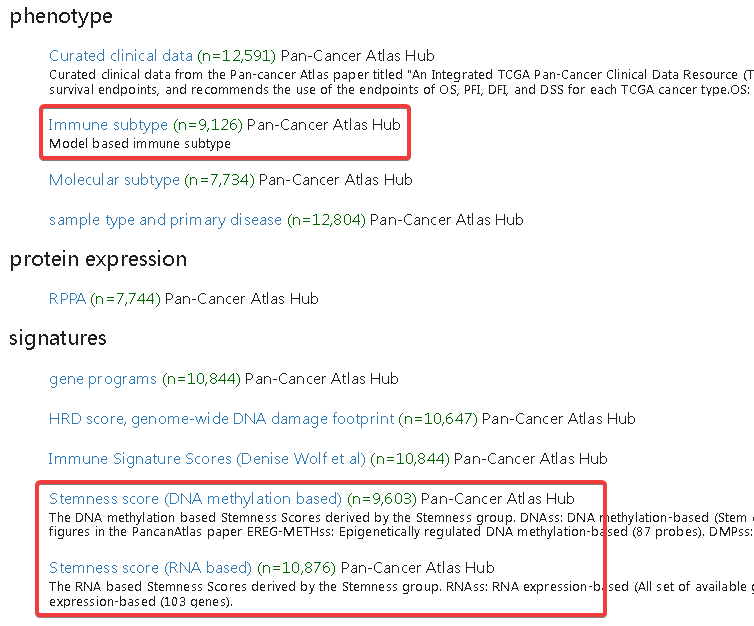
panGenes16.RNAssCor.R
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("limma")
#install.packages("corrplot")
#引用包
library(limma)
library(corrplot)
expFile="panGeneExp.txt" #表达输入文件
scoreFile="StemnessScores_RNAexp_20170127.2.tsv" #RNAss文件
scoreType="RNAss" #干细胞指数
#setwd("D:\\biowolf\\panGenes\\16.RNAssCor") #设置工作目录
#读取表达文件
exp=read.table(expFile, header=T,sep="\t",row.names=1,check.names=F)
exp=exp[(exp[,"Type"]=="Tumor"),]
#读取RNAss文件
STEM=read.table(scoreFile, header=T,sep="\t",row.names=1,check.names=F)
STEM=t(STEM)
#相关性检验
outTab=data.frame()
pTab=data.frame()
#按肿瘤类型循环
for(i in levels(factor(exp[,"CancerType"]))){
exp1=exp[(exp[,"CancerType"]==i),]
exp1=as.matrix(exp1[,1:(ncol(exp1)-2)])
row.names(exp1)=gsub(".$","",row.names(exp1))
exp1=avereps(exp1)
#样品取交集
sameSample=intersect(row.names(STEM),row.names(exp1))
STEM1=STEM[sameSample,]
exp1=exp1[sameSample,]
x=as.numeric(STEM1[,scoreType])
pVector=data.frame(i)
outVector=data.frame(i)
#按基因循环
genes=colnames(exp1)
for(j in genes){
y=as.numeric(exp1[,j])
corT=cor.test(x,y,method="spearman")
cor=corT$estimate
pValue=corT$p.value
pVector=cbind(pVector,pValue)
outVector=cbind(outVector,cor)
}
pTab=rbind(pTab,pVector)
outTab=rbind(outTab,outVector)
}
#输出相关性的表格
colNames=c("CancerType",colnames(exp)[1:(ncol(exp)-2)])
colnames(outTab)=colNames
write.table(outTab,file="RNAssCor.cor.txt",sep="\t",row.names=F,quote=F)
#输出相关性检验p值的表格
colnames(pTab)=colNames
write.table(pTab,file="RNAssCor.pval.txt",sep="\t",row.names=F,quote=F)
#RNAss相关性图形
pdf("RNAssCor.pdf",height=5,width=8)
row.names(outTab)=outTab[,1]
outTab=outTab[,-1]
corrplot(corr=as.matrix(t(outTab)),
title=paste0("\n\n\n\n",scoreType),
col=colorRampPalette(c("blue", "white", "red"))(50))
dev.off() 
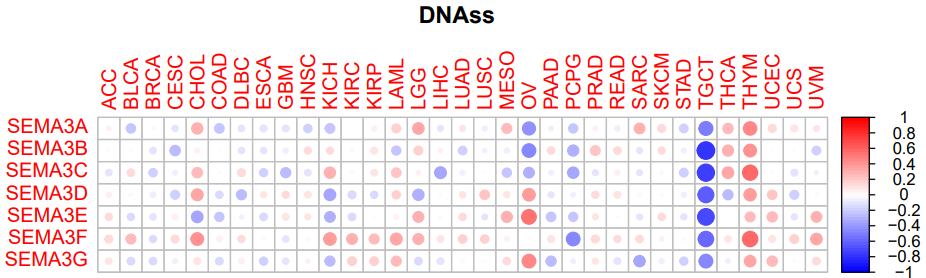
五、肿瘤干细胞相关性(DNAss)
需要使用StemnessScores_DNAmeth_20170210.tsv
panGenes17.DNAssCor.R
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("limma")
#install.packages("corrplot")
#引用包
library(limma)
library(corrplot)
expFile="panGeneExp.txt" #表达输入文件
scoreFile="StemnessScores_DNAmeth_20170210.tsv" #DNAss文件
scoreType="DNAss" #干细胞指数
#setwd("D:\\biowolf\\panGenes\\17.DNAssCor") #设置工作目录
#读取表达文件
exp=read.table(expFile, header=T,sep="\t",row.names=1,check.names=F)
exp=exp[(exp[,"Type"]=="Tumor"),]
#读取DNAss文件
STEM=read.table(scoreFile, header=T,sep="\t",row.names=1,check.names=F)
STEM=t(STEM)
#相关性检验
outTab=data.frame()
pTab=data.frame()
#按肿瘤类型循环
for(i in levels(factor(exp[,"CancerType"]))){
exp1=exp[(exp[,"CancerType"]==i),]
exp1=as.matrix(exp1[,1:(ncol(exp1)-2)])
row.names(exp1)=gsub(".$","",row.names(exp1))
exp1=avereps(exp1)
#样品取交集
sameSample=intersect(row.names(STEM),row.names(exp1))
STEM1=STEM[sameSample,]
exp1=exp1[sameSample,]
x=as.numeric(STEM1[,scoreType])
pVector=data.frame(i)
outVector=data.frame(i)
#按基因循环
genes=colnames(exp1)
for(j in genes){
y=as.numeric(exp1[,j])
corT=cor.test(x,y,method="spearman")
cor=corT$estimate
pValue=corT$p.value
pVector=cbind(pVector,pValue)
outVector=cbind(outVector,cor)
}
pTab=rbind(pTab,pVector)
outTab=rbind(outTab,outVector)
}
#输出相关性的表格
colNames=c("CancerType",colnames(exp)[1:(ncol(exp)-2)])
colnames(outTab)=colNames
write.table(outTab,file="DNAssCor.cor.txt",sep="\t",row.names=F,quote=F)
#输出相关性检验p值的表格
colnames(pTab)=colNames
write.table(pTab,file="DNAssCor.pval.txt",sep="\t",row.names=F,quote=F)
#DNAss相关性图形
pdf("DNAssCor.pdf",height=5,width=8)
row.names(outTab)=outTab[,1]
outTab=outTab[,-1]
corrplot(corr=as.matrix(t(outTab)),
title=paste0("\n\n\n\n",scoreType),
col=colorRampPalette(c("blue", "white", "red"))(50))
dev.off()